
5.03: B. Other Allosteric Proteins
- Page ID
- 158385


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Fundamental Concepts of Allostery
- Define allosterism and distinguish between allosteric binding at orthosteric versus allosteric sites.
- Explain how environmental factors (ligands, substrates, ions, and other effectors) influence protein conformation and cooperativity.
-
Models of Allosteric Regulation
- Describe the Monod-Wyman-Changeux (MWC) model and its application to diverse protein systems beyond hemoglobin.
- Compare and contrast the MWC model with other models (e.g., sequential/KNF model) in explaining allosteric transitions.
-
Multisubunit Enzymes and Allosterism
- Analyze how multimeric enzymes (such as lactate dehydrogenase and aspartate transcarbamylase) utilize allosteric transitions (T ↔ R states) to modulate catalytic activity.
- Interpret kinetic and binding curves (including sigmoidal versus hyperbolic plots, and double-reciprocal plots) to evaluate enzyme behavior under varying substrate and effector concentrations.
-
Structural Insights through Molecular Visualization
- Utilize interactive 3D molecular models (via tools such as iCn3D) to examine conformational changes between the T and R states in enzymes and ion channels.
- Identify key structural elements (e.g., subunit interfaces, allosteric binding pockets) that facilitate cooperative transitions in multisubunit complexes.
-
Allosterism in Diverse Protein Systems
- Explore examples of allosterism in systems beyond classical enzymes, including ligand-gated ion channels and structural viral proteins.
- Discuss how conformational shifts in viral capsids and ion channels contribute to their function (e.g., ion transport, viral assembly, and maturation).
-
Comparative Allosterism: Homotropic vs. Heterotropic Effects
- Differentiate between Type I (homotropic) allosterism, where a substrate modulates its own binding across multiple sites, and Type II (heterotropic) allosterism, where distinct effectors (inhibitors or activators) modulate substrate affinity.
- Evaluate experimental approaches used to characterize these different modes of allosterism (e.g., varying substrate vs. effector concentration in kinetic assays).
-
Integration of Allosteric Regulation in Cellular Contexts
- Examine how allosteric regulation of key enzymes (like phosphofructokinase and lactate dehydrogenase) impacts metabolic pathways and cellular homeostasis.
- Understand the broader implications of allosterism in pharmacology, including the development of drugs that target allosteric sites (e.g., the HIV capsid inhibitor lenacapavir).
-
Allosterism in Monomeric Proteins
- Investigate how monomeric enzymes, which possess multiple binding sites, can also exhibit allosteric behavior.
- Analyze structural and kinetic evidence for conformational changes in monomeric proteins (e.g., RecA and thrombin) that modulate their activity.
By achieving these learning goals, students will develop a comprehensive understanding of how allosteric regulation underpins the function of a wide range of protein complexes, linking structural transitions to catalytic and regulatory outcomes in diverse biological contexts.
Allosterism in other multisubunit protein complexes
Changeux (of the MWC model) has written eloquently about the occurrence and effects of allostery in other proteins. We will encounter these proteins in other chapters, but we will present some here before the chapter in which they are usually discussed. We do this to show that other proteins display allostery and that the MWC can often be used to describe their behaviors. This offers a rationale to discuss allosterism using hemoglobin with its nonstandard covalent ligands as a model for allosteric binding proteins and enzymes.
Environmental factors such as ligands and allosteric modulators can shift the degree of cooperativity for ligand binding, promote allosteric rearrangements, and T ↔ R transitions of proteins other than hemoglobin. We offer several examples of multimeric proteins (complexes) that display allosterism. Many of these allosteric proteins not only bind ligands but also act as catalysts. One protein, a ligand-gated ion channel, moves ions across a membrane. Others catalyze the chemical transformation of a substrate to a product. Another is a structural viral protein. The examples involving catalysis are more complex since an additional step (transport of ions or alteration in covalent bonds) after binding affects protein function. This extra step can be described as a rate, so we explore rate vs ligand concentration, not just fractional saturation vs ligand concentration curves.
Lactate dehydrogenase (LDH)
LDH is an enzyme that catalyzes the reversible reduction of the 3-carbon carboxylic acid pyruvate to lactate by the reducing agent NADH, as shown in the reaction below. The enzyme's name is descriptive of the reverse reaction in which lactate is oxidized.
pyruvate + NADH + H+ ↔ lactate + NAD+
Its activity is modulated by the allosteric activator fructose 1,6-bisphosphate (FBP). The kinetics can be modeled using the MWC model, in which the enzyme exists in T (tense/taut) and R (relaxed) allosteric states. FBP binds preferentially to the R state.
Figure \(\PageIndex{20}\) shows an interactive iCn3D model comparing the T state of bacterial L-lactate dehydrogenase with bound NAD+ from Bifidobacterium longum (1LLD), and the R state of the enzyme from Geobacillus stearothermophilus (2LDB) with bound NAD+ and the allosteric activator fructose 1,6-bisphosphate (F6P). Toggle between the two states using the "a" key.

 Figure \(\PageIndex{20}\): Comparison of the T (1LLD) and R (2LDB) states of bacterial L-lactate dehydrogenase with bound NAD+ and allosteric activator F6P (in R state) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...YEXKSp5J1ErFT6
Figure \(\PageIndex{20}\): Comparison of the T (1LLD) and R (2LDB) states of bacterial L-lactate dehydrogenase with bound NAD+ and allosteric activator F6P (in R state) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...YEXKSp5J1ErFT6
The enzyme's substrate NAD+, the allosteric activator F6P for the R state, and SO42- (from ammonium sulfate used to crystallize the protein) are shown in spacefill and labeled.
Aspartate transcarbamylase (ATCase)
This enzyme catalyzes the addition of aspartate and carbamoyl phosphate to form carbamoyl aspartate, the first step in the pathway for the synthesis of the pyrimidine nucleotides cytidine triphosphate (CTP) and uridine triphosphate (UTP).
The end products of the pathway, CTP and UTP, feed back and allosterically inhibit the enzyme. In contrast, ATP is an allosteric activator. This prevents a buildup of pyrimidine nucleotides over purine nucleotides since equal amounts are needed for nucleic acid synthesis.
Figure \(\PageIndex{21}\) shows an interactive iCn3D model comparing the T (4FYW) and R (1D09) states of asparatate transcarbamylase (ATCase). Toggle between the two states using the "a" key.
oR(1D09)State.png?revision=1&size=bestfit&width=422&height=382)
Figure \(\PageIndex{21}\): Comparison of the T tense (4FYW) and R (1D09) relaxed state of aspartate transcarbamylase (ATCase). Toggle between the two states using the "a" key. (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...MHLtC9ALwWVia6
Each of the subunits is shown in a different color. The T state (4FYW) has bound CTPs (at the periphery, shown in spacefill), while the R state has a bound substrate analog, N-(phosphonacetyl)-L-aspartic acid. High levels of the substrate (or substrate analog) shift the equilibrium to the active R state.
Pentameric ligand-gated ion channels (bacterial)
Protein channels in membrane bilayers are needed to "catalyze" and regulate the flow of ions across the hydrophobic membrane. Hence, it makes sense that channels exist in closed and open states. One example is the bacterial GLIC pentameric ligand-gated ion channel, opened by ligand binding, often called ligand-gating.
Figure \(\PageIndex{22}\) shows an interactive iCn3D model comparing the GLIC pentameric Ligand-Gated Ion Channel Loop2-22' oxidized mutant in a locally-closed conformation (LC3 subtype) (3TLV) and the A237F mutant channel in the open conformation (3LSV). Toggle between the two states using the "a" key.
%25C2%25A0to_the_open_conformation_(3LSV).png?revision=1&size=bestfit&width=288&height=359)
Figure \(\PageIndex{22}\): Comparison of the GLIC pentameric Ligand-Gated Ion Channel Loop2-22' oxidized mutant in a locally-closed conformation (LC3 subtype) (3TLV) and the A237F mutant of the pentameric ligand-gated ion channel from Gloeobacter Violaceus in the open conformation (3LSV). Toggle between the two states using the "a" key. (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...GKiPY9Jh7tQXTA
Note that the outer (red) and inner (blue) membrane leaflets are shown only in the closed channel (3TLV). The green spheres represent chloride ions.
Nudaurelia capensis ω virus capsid
This hallow viral protein structure surrounds the internal viral genome, so it is an example of allostery in a protein complex that is neither a transporter nor an enzyme. This hetero 480-mer with icosahedral symmetry changes its global shape when the immature capsid undergoes selective and limited proteolysis to form the mature capsid, as illustrated in Figure \(\PageIndex{22}\).

The R form is more open. This is a wonderful example of the global conversion of all subunits from a "T" to an "R" state, which is necessary in this case to preserve the exquisite symmetry!
Most drugs used to treat AIDS and other viruses target the active sites of viral enzymes. A breakthrough drug to treat HIV infections has been developed. Lenacapavir, binds to the HIV capsid proteins surrounding and protecting the interior viral RNA genome. The structure of the drug is shown below.

Representations of the HIV capsid structures are illustrated in Figure \(\PageIndex{n1}\) below. The full capsid forms a cone similar to buckminsterfullerene. It is comprised of many copies of a monomer (CA), which is arranged into around 250 CA hexamer and 12 CA pentamer assemblies.
 |
 |
Figure \(\PageIndex{n1}\): Structure of the HIV capsid. Rossi, E.; Meuser, M.E.; Cunanan, C.J.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life 2021, 11, 100. https://doi.org/10.3390/life1102010. Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Left: A schematic of the HIV-1 virion. Envelope proteins, GP41 and GP120, surround the host-derived membrane surface, lined internally with a layer of matrix protein. Inside the virion are viral proteins and the CA core containing the HIV-1 genome and proteins essential for infection. Image created with BioRender.com
Right: HIV-1 CA monomers oligomerize into hexamers and pentamers that assemble to form the capsid fullerene-cone core. (A) Shows the CA monomer with the amino-terminus colored blue and the carboxyl-terminus colored tan (PDB 3H47). (B) Shows the CA hexamer with each subunit colored differently (PDB 3H47). (C) Shows the hexamer with the amino terminus of each subunit colored blue and the carboxyl terminus colored tan (PDB 3H47). (D) Shows the CA pentamer with each subunit colored differently (PDB 3P05). (E) Shows the pentamer with the amino terminus of each subunit colored blue and the carboxyl terminus colored tan (PDB 3P05). (F) The full capsid core structure contains approximately 250 hexamer oligomers and 12 pentamer oligomers (PDB 3J3Q). Image created with the PyMOL Molecular Graphics System, Version 2.4 Schrödinger, LLC.
The journal Science named it the science breakthrough of 2024. Efficacy trials show that it reduces the chance of getting AIDs by an astonishing 99.9%. The drug is injected since it is very hydrophobic. Luckily, it has a very long lifetime in the blood, so only 2 injections per year are required. It appears to block HIV entry into cells by blocking sites on the capsid that interact with human proteins. Once it enters the cell, the virus must pass through the nuclear membrane to deliver the viral RNA genome. Lanacapavir appears to increase the cone's rigidity, so it has great difficulty crossing into the nucleus, which requires conformational flexibility.
Figure \(\PageIndex{n2}\) shows an interactive iCn3D model of the HIV capsid hexamer bound to Lenacapavir (GS-6207) (PDB ID 6V2F)
_(PDB_ID_6V2F).png?revision=1&size=bestfit&width=547&height=450)
Figure \(\PageIndex{n2}\): HIV capsid hexamer bound to Lenacapaviar (GS-6207) (PDB ID 6V2F) (Copyright; author via source). Click the image for a popup or use this external link: https://www.ncbi.nlm.nih.gov/Structu...Ym6iAaHLe36hV9
The six-subunit assembly has C6 symmetry, so it can be rotated 360/6 degrees to reproduce the structure. The noncovalent interactions of one lenacapavir with the hexamer are shown in detail. Similar drugs might be developed to treat other viral diseases.
Figure \(\PageIndex{n2}\) shows an interactive iCn3D model of the Mature HIV-1 capsid structure (3J3Q, long load time) that shows the conical structure of the capsid assembly.
.png?revision=1&size=bestfit&width=231&height=378)
Figure \(\PageIndex{n2}\): Mature HIV-1 capsid structure (3J3Q). (Copyright; author via source). Click the image for a popup or use this external link:https://www.ncbi.nlm.nih.gov/Structu...5SkLEzEs8U7Qm9
Here is a link to an amazing animation by Janet Iwasa et al. that shows how Lenacapavir works.
Now, look at the binding and rate curves for some multimeric allosteric enzymes. Since this is all a bit complicated, let's review again the difference between what we call Homotropic or Type 1 and Heterotropic or Type II allosterism:
Homotropic or Type I: Increasing the amount of a substrate can induce conformational changes in a multisubunit protein to a form that has apparently higher (or potentially lower as well) affinity for the substrate in the remaining unoccupied substrate binding sites. In this case, the substrate is binding to the orthosteric site. These sites are where substrates bind but also competitive inhibitors (if the protein is an enzyme) and agonists or competitive antagonists of receptors. We will explore enzymes and receptors later in this book. In Homotropic or Type I allosterism, binding or kinetic curves show sigmoidal fractional saturation (or kinetic curves) with increasing substrate concentrations.
Heterotropic or Type II: Increasing amounts of a chemical species (an inhibitor or activator) can bind to an allosteric site, affecting the substrate's binding to the orthosteric site. The regulators shift and change the shape of the Y or rate curves vs substrate. In experiments to show this kind of allosterism, you wouldn't change the substrate and allosteric effector concentrations simultaneously since the resulting data and graphs would be hard to interpret. You could change the ligand or substrate that binds to the orthosteric site over a large range of concentrations (hopefully over a 1000 - 10,000 fold change or 4 log units) in several different experiments, with each experiment having a different fixed concentration of the allosteric effector. Alternatively, you could perform the experiment over a large concentration range of a given allosteric effector (again, a 1000-10,000 fold change if possible) in several different fixed concentrations of ligand or substrate in a series of experiments.
Rate vs ligand curves for allosteric proteins that catalyze chemical reactions
Since we have already seen an example of homotropic or Type I allosteric binding curves (hemoglobin binding dioxygen), let's look at a few examples of heterotropic or Type II allosteric binding in multisubunit proteins, since their graphs are a bit more complicated. We realize the curves below show relative rates of enzymes and not relative fractional saturation of enzymes, but the same principles apply.
Phosphofructokinase
Figure \(\PageIndex{23}\) shows an example of allosteric kinetic (not just binding) curves for Phosphofructokinases A (Pfk A) and B (Pfk B) from Mycobacterium tuberculosis. The enzyme catalyzes the phosphorylation of fructose-6-phosphate (F6P) by ATP to produce fructose-1,6-bisphosphate (F1,6-BP) and ADP.
F6P + ATP → F1,6-BP + ADP

Figure \(\PageIndex{23}\): The dependence of Pfk A and Pfk B activities on the concentration of Mg2+. Individual reactions were performed in buffers containing fixed initial concentrations for both substrates (1 mM F6P and ATP) with the concentration of Mg2+ varied. Snášel, J. et al. Int. J. Mol. Sci. 2021, 22, 1483. https://doi.org/10.3390/ijms22031483. Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Only Pfk A shows allosteric activation of the enzyme by Mg2+, run under fixed initial (and probably saturating) concentrations of the substrates F6P and ATP. The Hill coefficient is 3.3 for Pfk A, which suggests that Mg2+ is important in maintaining/promoting the active site and formation of the enzyme tetramer. Pfk B shows hyperbolic kinetics and no allosterism, with a Hill coefficient close to 1. These curves are modeled with the Hill equation and not the MWC equation.
Lactate Dehydrogenase
Again, this enzyme catalyzes the following reaction:
pyruvate + NADH + H+ ↔ lactate + NAD+
The graphs in Figure \(\PageIndex{24}\) show relative inhibition (graph A, top) and double-reciprocal plots (C, bottom) for the enzyme lactate dehydrogenase B (LDHB) in the presence of an allosteric inhibitor, AXKO0046. This enzyme catalyzes the reduction of pyruvate by NADH (the substrate) to form lactic acid and NAD+ (the products). The sigmoidal nature of the graphs is very clear (top panel).
![Graphs showing NADH concentration effects on inhibition and kinetic analysis. Panel (a) displays inhibition percentages against AXKO-0046 concentration. Panel (c) presents kinetic plots of 1/v versus 1/[NADH].](https://bio.libretexts.org/@api/deki/files/94860/selective_inhibitor_of_human_lactateAllosterismFig3_V2.svg?revision=1&size=bestfit&width=474&height=712) |
 |
Figure \(\PageIndex{24}\): Biochemical characterization of AXKO-0046. LDHB inhibition by AXKO0046 was studied using varying concentrations of (a) NADH (c) Double reciprocal (Lineweaver-Burk) plots of the kinetic data. Shibata, S. et al. Sci Rep 11, 21353 (2021). https://doi.org/10.1038/s41598-021-00820-7. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
For a more detailed analysis, click below if you have already studied enzyme kinetics.
- Link
-
These graphs are a bit more complicated since, in this case, the initial concentration of one substrate is varied while the other concentration is fixed (in contrast to the PfkA experiments in which the initial concentrations of both substrates were held constant).
Let's look at graph C (bottom) first. Instead of showing graphs of rate vs [NADH], the authors showed double-reciprocal kinetic plots, run with varying [NADH] and at one fixed concentration of pyruvate (not given) and several fixed concentrations of inhibitor. The graphs look like they are generally straight lines except at the end, which occurs at a low concentration of NADH (which gives the highest value of 1/[NADH] = 0.10). At this point, 1/rate (given as 1/v) data points are higher than the best-fit line would suggest, implying that the rate v is "abnormally" low. That rate accelerates as [NADH] increases (as 1/[NADH] decreases in a manner consistent with allosterism. This suggests that at any given fixed concentration of inhibitor and fixed pyruvate concentration, the graphs of rate vs substrate (NADH) (i.e., not the double-reciprocal plot) would be sigmoidal. It differs from the PfkA graph (Fig. 23), in which the x-axis variable is the allosteric activator Mg2+.
Look at Graph A (top), which uses varying inhibitor concentrations and several fixed concentrations of reactant NADH. This is analogous to the graph for PfkA, but notice that on the x-axis, the log [inhibitor] is plotted instead of the [inhibitor]. These are NOT plots of v vs. [substrate], expected to be hyperbolic, or v vs. log[substrate], which is expected to be sigmoidal. (A lesson here is to look carefully at the axes). But note something unusual about the curves. The plateau for inhibition is not the same at each NADH concentration. The highest % inhibition (red and purple curves) occurs when the substrate [NADH] is highest. (We will see in the next chapter that this is a sign of what is called uncompetitive inhibition).
X-ray crystal structures show that the inhibitor (AXKO-0046) does bind to an allosteric site, not the active orthosteric site. It appears to bind in the interface of the LDHB tetramer. The graphs show that over four orders of magnitude of inhibitor (4 log units), the inhibition goes from 0 to about 100%. This is expected if the sigmoidal semi-log curves gave hyperbolic curves with [inhibitor] plotted on the x-axis.
This may seem confusing, but such sigmoidal curves are found in plots of rate vs log concentration of allosteric activators and inhibitors, as discussed in Chapter 5.1. So, don't immediately conclude that a sigmoidal curve implies allosterism. Look at the reactions and relative concentrations carefully.
Consider this example. What if a protein binds a ligand L and an inhibitor I at the same orthosteric site? If one bound, the other couldn't. This is an example of a classical competitive, non-allosteric inhibition. Now, what if an inhibitor, I, binds to an allosteric site, and when bound, it alters the conformation of the orthosteric site such that the ligand cannot bind? The binding of L and I would be mutually exclusive. This would produce the same binding curves as the classical competitive inhibition. In either case, at very high ligand concentrations, the effect of the inhibitor would be lost, and full maximal binding would be observed. It would just take higher concentrations of ligand to get the same fractional saturation of the protein in the presence of the inhibitor than in its absence. In the presence of a fixed concentration of these competitive inhibitors, the effective KD would be higher. Y vs L curves for both would be hyperbolic, and double-reciprocal plots would be linear.
Allostery within monomeric protein
Allosterism can also occur in monomeric proteins:
Type I (again, our nomenclature) allosterism can occur in monomers that have more two or more binding sites for a ligand/substrate, and if the binding of ligand/substrate to one site significantly alters the affinity of the other site for substrate enough to produce a nonhyperbolic, sigmoidal binding/kinetic curve for substrates. This case is different than the binding of substrate to two different preformed substrate binding sites, each with a different fixed affinity, which we discussed in Chapter 5.1 (scroll down to binding of a ligand to two independent sites). Again, we show the graph of fractional saturation Y vs. L for the binding of a ligand to two preformed sites of different affinities below.
Note that the above graph doesn't look sigmoidal. It is essentially hyperbolic except in the extreme case when one of the KD is much less than the other, AND at low ligand concentration such that the higher affinity binding leads to an abrupt titration curve-like saturation of the low KD site before the second site has much occupancy.
Rec A is an OK example of a "possible" Type I allosteric monomer binding protein (if you have a better example, let us know!). This protein is required for homologous recombination in bacteria. It has ATPase activity and catalyzes ATP-driven homologous pairing and strand exchange of DNA required for DNA repair. The structure is known for the Mycolicibacterium smegmatis apo form of the enzyme, the enzyme:substrate (dATP, a substrate analog) complex, and the enzyme:substrate:allosteric effector (a second dATP and possibly citrate) complex.
The enzyme has three domains (N-terminal 1-30, the major M domain (31-269), and the C-terminal (270-349). The M domain is the catalytic domain, which has nucleotide triphosphate hydrolase activity. It binds nucleotides and DNA and interacts with the N domain of another RecA to promote the polymerization of RecA into a filament. The C-terminal domain is disordered but becomes ordered when bound to a second dATP in the crystal structure.
Figure \(\PageIndex{25}\) shows conformational changes in RecA:dATP (the ES complex) on binding a second dATP (the ESA complex), where A is the likely allosteric activator (the second bound dATP). A result of this ordering on binding is likely the polymerization of the RecA into filaments.
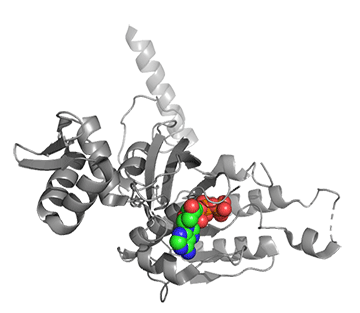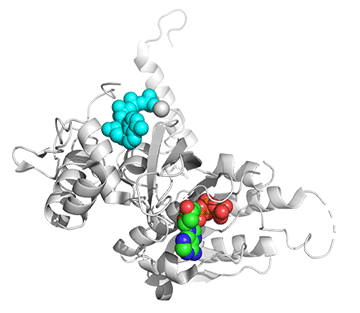
Figure \(\PageIndex{25}\): Conformational changes in RecA:dATP (the ES complex) on binding a second dATP (the ESA complex).
The dATP in the catalytic site is shown in spacefill with CPK colors. The ES complex is a darker gray protein with one bound dATP (spacefill, CPK colors). The ESA complex is shown in lighter gray with dATP bound in the catalytic (orthosteric) site in CPK colors and a second dATP (spacefill, cyan) bound in the putative allosteric site in the C domain.
Type II: increasing amounts of a chemical species (an inhibitor or activator) that binds to an allosteric site in a monomeric protein could affect the binding of the substrate to an orthosteric site in the monomer. In this case, as in Type II for multimeric proteins, you could again run two different types of experiments (one with varying substrate at 3-4 different fixed allosteric effector concentrations or vice/versa.
One example is thrombin, the last protease in a cascade of clotting proteins. The proteins are synthesized as inactive precursors (zymogens) that become activated on limited proteolysis. Active thrombin is a procoagulant enzyme that cleaves circulating fibrinogen (and other procoagulant molecules) into fibrin. This then self-associates to form a fibrin clot.
Paradoxically, thrombin also has anticoagulant properties. It can cleave another circulating protein, Protein C, which inhibits further clotting. Thrombin does so when it binds a transmembrane protein, thrombomodulin, present in the plasma membrane of the endothelial cells that line blood vessels.
These contrasting activities support the notion that thrombin has two interconverting conformations, each stabilized by different ligands or proteins. One such ligand is the simple monatomic ion Na+. Indeed, thrombin appears to have two main catalytic conformations: a high-activity “fast” form (with bound Na+) and a low-activity “slow” form (without bound Na+). The fast form with bound Na+ (15 Å from the active site) appears to be the procoagulant form, while the slow form is the anticoagulant form.
Figure \(\PageIndex{26}\) shows an interactive iCn3D model comparing the anticoagulant slow form of thrombin (1SGI) and the procoagulant sodium-bound fast form of thrombin (1SG8). Toggle between the two states using the "a" key.
__procoagulant_sodium-bound_fast__thrombin_(1SG8).png?revision=1&size=bestfit&width=228&height=241)
Figure \(\PageIndex{26}\): Anticoagulant slow form (1SGI) and the procoagulant sodium-bound fast form of thrombin (1SG8). Toggle between the two states using the "a" key. (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...kxnATvMdVD1566
The magenta represents the slow form, and the cyan with bound Na+ is the fast form which has enhanced coagulant activity
Table \(\PageIndex{1}\) below shows a list of monomeric allosteric proteins and their PDB file codes. The proteins (P) are enzymes that bind a substrate (S) and an allosteric effector (A) to form PS, PA, PAS complexes (adapted from Wang et al. J. Phys. Chem. Lett. 2021, 12, 5404−5412)
| protein | P | PA | PS | PAS | Effect |
| protein RecA (RecA) | 2OES | 2ODN | 2G88 | activation | |
| mitogen-activated protein kinase 8 (MAP8) | 1UKH | 3O2M | 2XRW | inhibition | |
| cAMP-dependent protein kinase catalytic. subunit alpha (Prkaca) | 4NTS | 4NTT | 4IAF | inhibition | |
| cAMP-dependent protein kinase catalytic. subunit alpha (Prkaca) | 4NTS | 1BKX | 4DG0 | activation | |
| cyclin-dependent kinase 2 (CDK2) | 3PXR | 3PXF | 1HCK | inhibition | |
| casein kinase II subunit alpha (CK2α) | 5ZN5 | 3H30 | 2PVR | inhibition | |
| myosin-2 heavy chain (mhcA) | 1FMV | 2JJ9 | 2JHR | inhibition | |
| tyrosine-protein phosphatase. nonreceptor type 1 (PTP1B) | 4QBW | 1T49 | 1PTV | inhibition | |
| 1T48 | |||||
| 1T4J | |||||
Summary
Chapter Summary: Allosterism in Multisubunit Protein Complexes
This chapter expands the concept of allosterism beyond hemoglobin to illustrate how a wide range of multisubunit proteins regulate their activity through ligand-induced conformational changes. The discussion emphasizes that environmental factors—including substrates, inhibitors, activators, and ions—can shift the equilibrium between different conformational states (commonly termed T [tense] and R [relaxed] states) in both catalytic and non-catalytic proteins.
Key Themes and Concepts:
-
Fundamentals of Allosteric Regulation:
The chapter introduces allosterism as a fundamental mechanism by which proteins modulate their activity. Allosteric transitions often involve cooperative changes that affect ligand binding and catalytic rates. Two types of allosteric effects are defined:- Type I (Homotropic): Where binding of a substrate at one site influences the binding affinity at other identical (orthosteric) sites, leading to sigmoidal binding curves.
- Type II (Heterotropic): Where distinct molecules (inhibitors or activators) bind to separate allosteric sites and modulate the protein’s activity.
-
Multisubunit Enzymes and Ion Channels:
The MWC model is used to explain the T ↔ R transitions in enzymes such as lactate dehydrogenase (LDH) and aspartate transcarbamylase (ATCase), where binding of allosteric activators (like fructose 1,6-bisphosphate) or inhibitors (such as CTP) shifts the equilibrium to the active or inactive state. In parallel, examples of ligand-gated ion channels (e.g., the GLIC channel) illustrate how allostery regulates ion flow across membranes by converting between closed and open states. -
Allostery in Structural Complexes:
Allosteric regulation is also observed in non-enzymatic complexes, such as viral capsids. For instance, the structural transition from an immature to a mature capsid in the Nudaurelia capensis ω virus, and the innovative design of HIV capsid inhibitors like lenacapavir, highlight the therapeutic potential of targeting allosteric sites. -
Kinetic Analyses and Graphical Interpretations:
The chapter reviews kinetic data and binding curves (including sigmoidal plots and double-reciprocal Lineweaver-Burk plots) to demonstrate how allosteric effectors modulate enzyme activity. Comparisons between enzymes with clear allosteric behavior (such as phosphofructokinase A) and those with hyperbolic kinetics (like phosphofructokinase B) underscore the nuances in allosteric regulation. -
Allostery in Monomeric Proteins:
Although often associated with multisubunit complexes, allosteric regulation can also occur in monomeric proteins that harbor multiple binding sites. Examples include RecA, which displays conformational changes upon binding additional dATP molecules, and thrombin, which exists in fast (Na⁺-bound, procoagulant) and slow (unbound, anticoagulant) forms.
Conclusion:
This chapter reinforces that allosterism is a versatile regulatory strategy found across diverse protein complexes. By integrating structural data, kinetic analyses, and mathematical modeling, students gain a comprehensive view of how conformational transitions underpin the regulation of enzyme activity, ion transport, and viral assembly. This understanding lays the groundwork for exploring therapeutic interventions that target allosteric sites, further connecting molecular mechanisms with biomedical applications.