
4.2: Secondary Structure and Loops


( \newcommand{\kernel}{\mathrm{null}\,}\)
-
Understand Helical Structures:
- Explain the general concept of protein helices as repetitive structures stabilized by hydrogen bonds between backbone amide Hs and carbonyl Os.
- Describe the idealized geometries of different helical types—alpha (α), 3₁₀, and pi (π) helices—including parameters such as residues per turn (n), pitch, and the characteristic phi/psi angles.
- Compare and contrast the alpha helix with the 3₁₀ and pi helices in terms of stability, dimensions, and side chain orientation.
- Interpret helical wheel projections to evaluate amphipathic characteristics and the distribution of hydrophobic and hydrophilic residues along the helix.
-
Examine Beta Structures:
- Distinguish between parallel and antiparallel beta strands, and describe how hydrogen bonding patterns and dihedral angles contribute to the formation of beta sheets.
- Explain how beta strands form "pleated sheet" structures and how the alternating orientation of side chains contributes to sheet stability and overall protein function.
- Evaluate structural models and interactive 3D representations (e.g., using iCn3D) to visualize beta sheets and beta barrels.
-
Analyze Turns, Loops, and Linkers:
- Define and differentiate between various types of reverse turns (beta turns, gamma turns, and hairpins) that connect elements of regular secondary structure.
- Describe the characteristics of short connector regions (loops, linkers, and bends) and explain how they influence the overall folding and dynamics of a protein.
- Recognize how specific amino acid sequences, particularly those rich in glycine and proline, facilitate tight turns and loops in protein structures.
-
Explore Backbone Dihedral Angles and Ramachandran Plots:
- Define the phi (φ) and psi (ψ) dihedral angles of the peptide backbone and explain how these angles determine the conformational space available to a protein.
- Interpret Ramachandran plots to identify the allowed and disallowed regions of φ/ψ space, and correlate these with common secondary structures (α-helices, β-sheets, and turns).
- Discuss how deviations in dihedral angles, due to side chain sterics or flexibility (as in glycine), affect the overall protein structure.
-
Assess Amino Acid Propensities in Secondary Structures:
- Describe the factors that determine the propensity of different amino acids to form specific secondary structures, using examples such as the Chou-Fasman parameters.
- Compare the roles of amino acids with or without beta-carbon branching in stabilizing or destabilizing helical conformations.
- Explain how the intrinsic properties of amino acids influence the overall stability and folding of the protein, and how these properties can be used to predict structural elements from sequence.
-
Integrate Structural Models with Functional Implications:
- Relate the specific secondary structure elements (helices, beta sheets, turns) to the overall 3D architecture and functional properties of proteins.
- Discuss how secondary structures contribute to the formation of tertiary and quaternary structures and ultimately influence protein activity and interactions.
- Utilize interactive modeling tools (e.g., iCn3D) to explore protein structures such as hydroxynitrile lyase (5Y02) and assess the spatial arrangement of secondary structure elements.
These learning goals are intended to provide students with both a conceptual and practical understanding of protein secondary structure, enabling them to analyze conformational features from both a theoretical and structural perspective.
Secondary structures are those repetitive structures involving hydrogen bonds between amide Hs and carbonyl Os in the protein backbone. These include
- helices (alpha - α, 310, and pi - π), in which the hydrogen bonds are within a short continuous stretch of amino acids (a strand),
- beta strands (β sheets) in which the hydrogen bonds are between backbone atoms (again amide Hs and carbonyl Os) on noncontinuous stretches of the protein and
- reverse turns occur within a very short, continuous stretch of amino acids.
Helices
A schematic showing idealized geometries of helices, with amino acids shown as dots for simplicity, is shown in Figure 4.2.1.

The pitch (p) represents the spacing between the chain on one side of the helix, the number of amino acids per turn (n), and handedness (+ = right-handed or – l= left-handed) are shown in the figure. There are three major types of helices in proteins: the alpha helix (n = 3.6), 310 helix (n = 3), and the pi helix (n = 4.4). Note that n is most commonly not an integer.
Alpha
The alpha helix is the most common type of helix. They are formed when the carbonyl O of the ith amino acid forms hydrogen bonds to the amide H of the ith+4 aa (4 amino acids away). Figure 4.2.2 shows a short section of an alpha helix running from the N-terminal (bottom) to the C-terminal (top) with the sequence DTASDAA. The amino acids i, i+1, ... i+4 are labeled at their alpha carbons. The red oval highlights the intrastrand H bond between the C=O of the ith amino acid (Asp) and the amide H of the ith+4 aa (Ser).
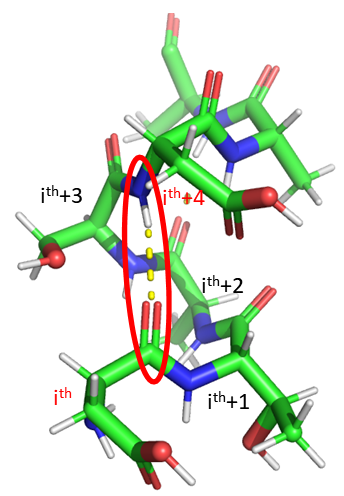
Figure 4.2.3 shows a cartoon image showing a longer helix and a schematic showing hydrogen bonding partners.
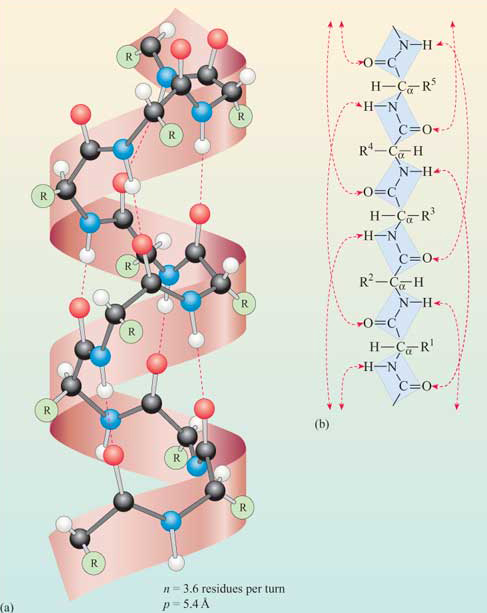
The phi/psi angles for amino acids in the alpha helix are approximately - 57,-47, which emphasizes the regular repeating nature of the structure. It can also be characterized by n (the number of amino acid units/turn = 3.6) and the pitch (the helix rise/turn = 5.4 angstroms = 0.54 nm). Since there are 3.6 amino acids per turn, and a full circle or turn is 3600, each amino acid is staggered at 1000 increments looking down on the helix axis. To refresh your mind, the phi/psi diagram for a fully extended polypeptide chain (phi 1800, psi 1800) is shown below in Figure 4.2.4.

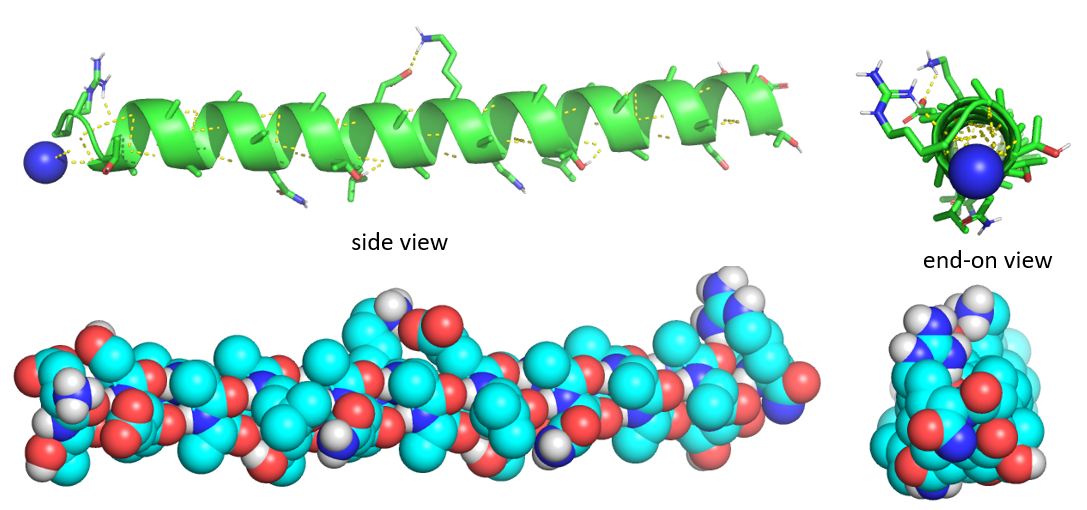
Figure 4.2.5 shows the side and end-on view of a helix from the antifreeze protein (1wfa) from the winter flounder. The green coil (often shown in red when displaying alpha helices in full proteins) shows the repetitive nature of the backbone. Note that side chains are pointing away from the helix axis. H-bonds are shown as yellow dotted lines within the backbone (one is also shown between two side chains on the top). The spacefill rendering is shown in colors that are optimal for colorblind people. The end-on view shows that the center of the helix is fully packed with atoms from the helix and is NOT open (a common misconception among students).
Some facts:
- the alpha helix is more compact than the fully extended polypeptide chain with phi/psi angles of 180o.
- in proteins, the average number of amino acids in a helix is 11, which gives three turns.
- the left-handed alpha helix, although allowed Ramachandran plot analyses, is never observed since the side chains are too close to the backbone.
- the core of the helix is packed tightly. No central cavities or pores are present in the helix.
- All the R-groups extend backward and away from the helix axis.
- Some amino acids are more commonly found in alpha helices. Amino acids can be divided into two kinds: those with branches at the beta C and those with none. Consider first those that aren't branched. Gly is too conformationally flexible to be found with high frequency in alpha helices, while Pro is too rigid. The amino acids with side chains that can H-bond (Ser, Asp, and Asn) and aren't too long appear to act as competitors of main chain H bond donors and acceptors and destabilize alpha helices. The rest with no branches at the beta C can form helices. Those with branches at the beta carbon (Val, Ile) destabilize the alpha helix due to steric interactions of the bulky side chains with the helix backbone. (Remember, left-handed alpha helices are not naturally found for similar reasons.)
- alpha keratins, the major component of hair, skin, fur, beaks, and fingernails, are almost all alpha helix.
Figure 4.2.6 shows an interactive iCn3D model of an alpha helix from bacteriophage T4 lysozyme (1DYG). Side chains, which are not involved in helix-stabilizing hydrogen bonds, are shown in cyan. H-bonds are shown as green dotted lines.
The amino acid side chain R-groups can be hydrophilic or hydrophobic. They can be localized in specific positions on the helix, forming amphipathic regions on the protein, or fully hydrophobic helices may extend through the plasma membrane as shown in Figure 4.2.7.
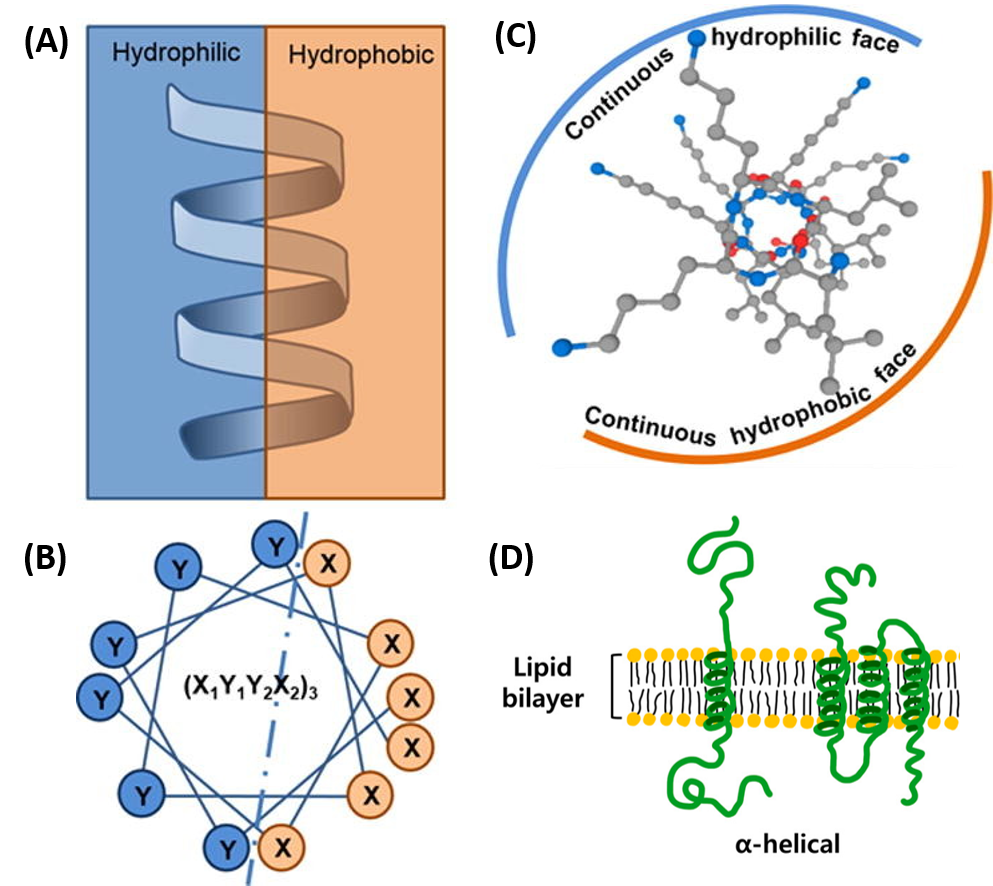
In amphipathic helices, hydrophilic residues are positioned on one side of the helix and hydrophobic on the other, as shown in the side view (A) or top-down views (B & C). R-groups may also be entirely hydrophobic within alpha helices that span the plasma membrane, as shown in (D).
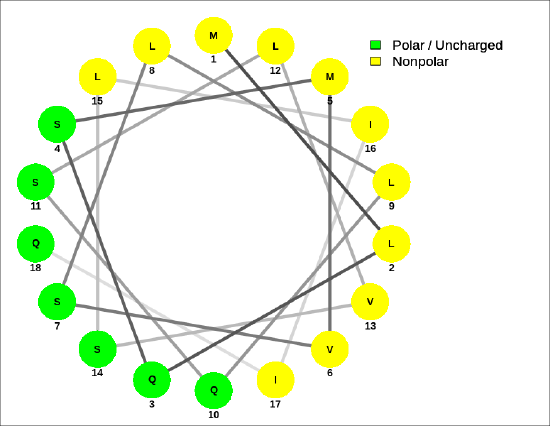
Helical wheel projections can show the polarity of the faces of the helix looking down the axis. Here are two such helical wheel projections:
For the sequence MLQSMVSLLQSLVSLIIQ, Figure 4.2.8 shows that the helix is amphiphilic.
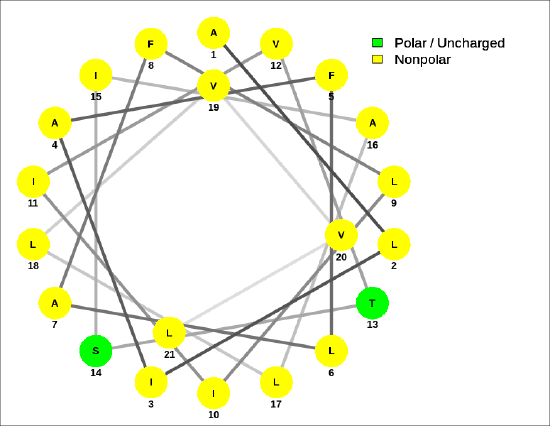
A helical wheel for the membrane-crossing section of the human receptor-type tyrosine-protein phosphatase C protein, ALIAFLAFLIIVTSIALLVVL, is shown in Figure 4.2.9.
The amide bond in the peptide has a significant permanent dipole moment. Since the dipoles of individual amino acids are oriented in the same direction in an alpha helix, the whole helix has a significant dipole moment. This is illustrated in Figure 4.2.10.
recreate this image:
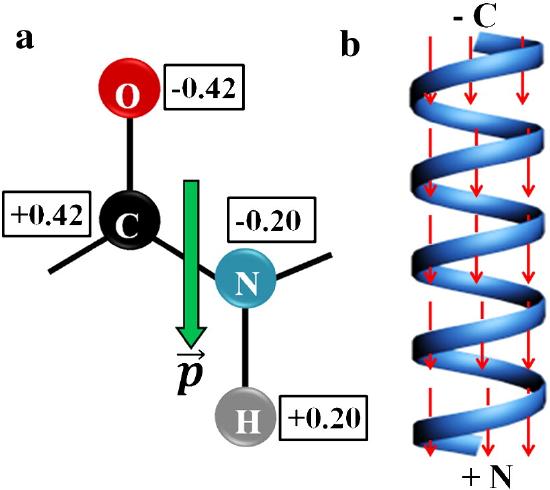
In the alpha helix, all the amides in the backbone point in one direction from the C-terminus to the N-terminus. This leads effectively to one long dipole of magnitude n(3.5) Debyes, where n is the number of amino acids in the helix. A long helix can produce a significant electric field and affect protein binding properties.
310 helices
The 310 helix is stabilized by hydrogen bonds between the carbonyl O of the ith amino acid and the amide H of the ith+3 aa (3 amino acids away). It has 3 residues/turn and a pitch (rise per turn) of 6 angstroms, with a rise of 1.3-2 angstroms/residue. Typical phi/psi angles are -500,-26°. As with the alternative description of the alpha helix, the 310 helix has 3 amino acids per turn and 10 atoms in the main chain/turn (counting Cα-N-C-Cα atoms). It is longer (for the same number of amino acids) and thinner. The amino acid side chains are staggered at 1200 increments as you look around the helix axis. Although not very prevalent (about 3 percent of protein amino acids are in 310 helix with an average of 3.3 amino acids in the helix), they presumably serve some function. 310 helices as long as 11 residues have been found.
A helix will be stable within a protein if the packing and noncovalent interactions favor it over other conformations. Many more alpha-helix side chain interactions with surrounding protein are likely, given a 1000 staggering of side chains compared to a 1200 staggering in a 310 helix, which lineup in 3 ridges looking down the helical axis. Molecular dynamics studies suggest that parts of a 310 helix might reversibly interconvert to an alpha helix, allowing conformational and binding flexibility. The S4 helix in some voltage-sensitive potassium ion channels with a canonical R1xxR2xxR3xxR4xxK5xxR6 (where R and K are Arg and Lys) have been shown to adopt a 310 helical conformation.
Figure 4.2.11 shows an interactive iCn3D model of a 9 amino acids (150-158) 310 helix from dienelactone hydrolase (1DIN)
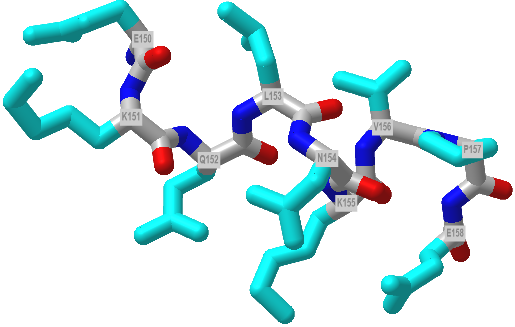
 Figure 4.2.11: A 310 helix from from dienelactone hydrolase (1DIN). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...Kufi7gmpSBdjYA (Copyright; author via source)
Figure 4.2.11: A 310 helix from from dienelactone hydrolase (1DIN). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...Kufi7gmpSBdjYA (Copyright; author via source)
π (pi) helices
This helix has 4.4 residues/turn and a helix pitch (rise) of about 4.1 nm angstroms. It has hydrogen bonds between the carbonyl O of the ith amino acid and the amide H of the ith+5 amino acids (5 amino acids away). Its rise is about 1.2 angstroms/residue and has approximate phi/psi angles of -550, -700. An alternative designation for the pi helix is 4.416, with 16 main chain atoms in 1 full turn (see above for the alpha helix). Some reasons for its low abundance include minimal contact between the main chain atoms, given the larger radius and phi-psi angles close to disallowed values. It might also not form kinetically as fast as the other helices since nucleation of it would be more difficult. At the same time, molecular dynamic simulation simulations show alpha helices can interconvert reversibly with pi helices. They are often found between two alpha helices, again suggesting dynamic interconversions between the two forms are likely.
About 55% of characterized pi helices contain five amino acids. Each side chain is staggered by 850, with a rise of about 1.3 Angstroms. Figure 12 below shows an interactive iCn3D model of a short pi helix (aa 265-276) from barley beta-D-glucan glucohydrolase (1x38).
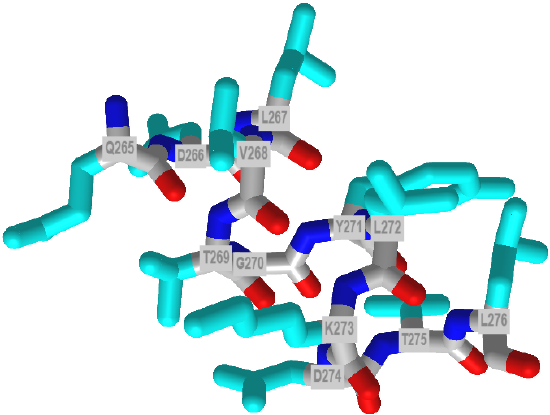
 Helices in Proteins: Comparison of alpha, 310, and pi helices
Helices in Proteins: Comparison of alpha, 310, and pi helices
Beta Structure
Beta Structure: Parallel and antiparallel beta strands are much more extended than alpha helices (phi/psi of -57,-47) but not as extended as a fully extended polypeptide chain (with phi/psi angles of +/- 180) as shown in the figure below. Parallel beta strands have phi/psi angles of -119, +113, while the antiparallel angles are -139, +135. Figure 4.2.13
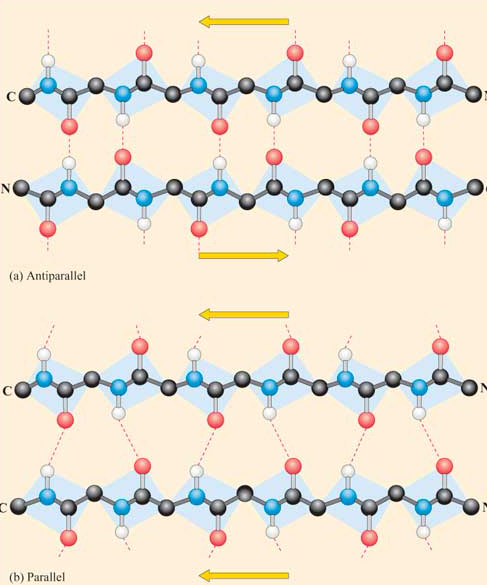
Each single strand of the beta-sheet can be pictured as a twofold helix, i.e., a helix with two residues/turn. The arrangement of each successive peptide plane is pleated due to the tetrahedral nature of the alpha C. Hydrogen bonds are inter-strand, not intra-strand, as in the alpha helix.
The figure below shows how the "pleats" in a sheet containing parallel beta strands can be envisioned as rippled sheets. Figure 4.2.14
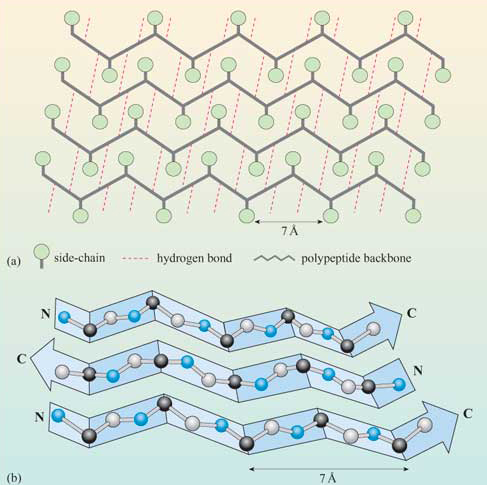
They can be visualized by folding a sheet of paper into narrow folds or pleated strips to make a "pleated sheet" of paper. Each strip of paper can be pictured as a single peptide strand in which the peptide backbone makes a zig-zag along the strip, with the alpha carbons lying at the folds of the pleats. The R groups are attached to the carbons and extend above and below the pleat folds in the trans conformation.
Consider a strand as a continuous, contiguous polypeptide backbone propagating in one direction. Hence, using this definition, a helix consists of a single strand, and all the H-bonds are within the strand (or intrastrand). A beta sheet would then consist of multiple strands since each "strand" is separated from other "strands" by an intervening contiguous stretch of amino acid, which bends within the protein in a way that allows the next section of the peptide backbone, the next "strand," to H-bond with the first "strand." But remember, even in this case, all the H-bonds holding the alpha and beta structures together are intramolecular.
In a parallel beta sheet structure, the optimal H bond pattern leads to a less extended structure (phi/psi of -119, +113) than the optimal arrangement of the H bonds in the antiparallel structure (phi/psi of -139, +135). Also, the H bonds in the parallel sheet are bent significantly. (i.e., the carbonyl O on one strand is not exactly opposite the amide H on the adjacent strand, as it is in the antiparallel sheet.) Hence, antiparallel beta strands are presumably more stable, although both are abundant in nature. Short parallel beta sheets of 4 strands or less are not common, which might reflect their lower stability.
The side chains in the beta sheet are normal to the plane of the sheet, extending out from the plane on alternating sides. Parallel sheets characteristically distribute hydrophobic side chains on both sides of the sheet, while antiparallel sheets are usually arranged with all the hydrophobic residues on one side. This requires alternating hydrophilic and hydrophobic side chains in the primary sequence. Antiparallel sheets are found in silk, with the sheets running parallel to the silk fibers. The following repeat is found in the primary sequence: (Ser-Gly-Ala-Gly)n), with Gly pointing out from one face and Ser or Ala from the other.
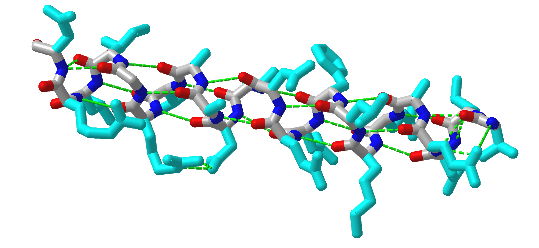
Unfortunately, no PDB structure of the silk "amyloid" protein shows this repetitive structure. The monomers and aggregates of this protein are quite insoluble, so few X-ray structures for proteins like this are available. Figure 4.2.15 shows an interactive iCn3D model of the N-terminal part (domain) of the Bombyx mori fibroin silk protein (pdb = 3UA0), which does give an excellent example of antiparallel beta sheets. Notice that two chains align to form a face of the curved antiparallel beta sheet.
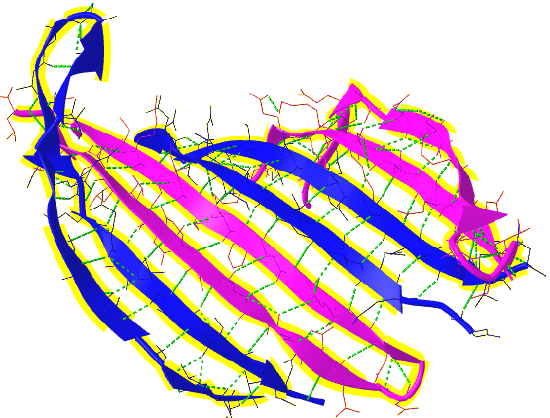
Figure 4.2.16 shows a more detailed static image of the antiparallel beta sheets in 3UA0. Note the yellow sticks between the strands representing the H-bonds.
Beta strands tend to twist in the right-hand direction. This leads to important consequences in how the beta strands are connected. Parallel strands can form twisted sheets, saddles, and beta barrels.
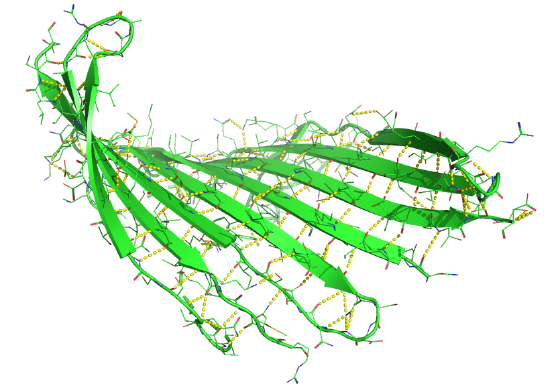
Figure 4.2.17 shows an interactive iCn3D model of the parallel beta sheet structure from the arabinose binding protein (1ABE).

Figure 4.2.18 shows a static image of the parallel beta sheets in 1ABE. Note the yellow sticks between the strands representing the H-bonds.
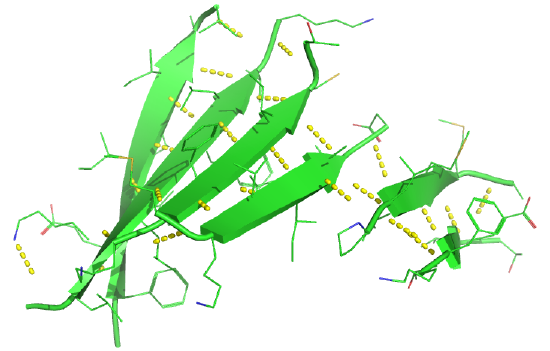
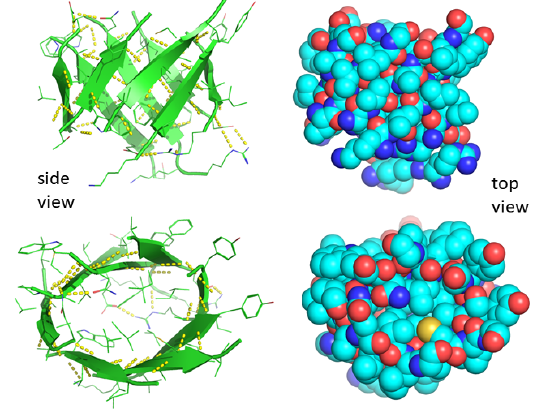
Figure 4.2.19 shows an interactive iCn3D model of a parallel beta barrel from the triose phosphate isomerase (1WYI).
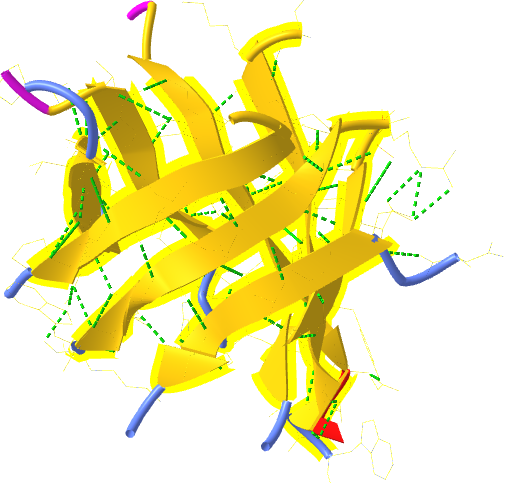
Figure 4.2.20 shows a static image of the parallel beta sheets in triose phosphate isomerase. Note the yellow sticks between the strands representing the H-bonds. Also, note that the barrel is not hollow but is filled with side chains.
Some facts on parallel beta structure:
- in parallel strands, right-handed connectivity is common.
- in a protein with parallel strands in a register, and given the inherent twist in the stands, the strands arrange to have the H bonds stretched equally at the ends of the chains, giving rise to a twisted saddle shape (top structure above).
- in a protein with parallel strands out of register, and given the inherent twist in the stands, the strands arrange to have the H bonds stretched equally at the ends of the chains, giving rise to a beta barrel (bottom structure above).
Connectors, Loops, Linkers, and Bends
About 50% of the amino acids in a globular (spherical) protein are in regular secondary structure (alpha or beta). The amino acids in helices and beta strands are connected by stretches of amino acids, which still have order. Still, that order is less regular than those found in helices and beta strands, characterized by stretches of amino acids with the same phi/psi angles. Some bear the hallmarks of secondary structure - intrachain hydrogen bonds hold them together. We will consider a few here.
Turns, Reverse Turns, and Hairpins
One example of a connector involving secondary structure (i.e. hydrogen bonds between amide Hs and carbonyl Os of the backbone), is a reverse turn called the beta bend or beta turn. These turns often connect successive antiparallel beta strands and are then called beta hairpins. A hairpin is a special case of a turn in which the direction of the protein backbone reverses, and the flanking secondary structure elements interact. For example, the beta hairpin connects two hydrogen-bonded, antiparallel β-strands. The word beta can be confusing. It does not mean the structure has hydrogen-bonded amino acids with the same phi-psi angles as beta strands. It's easy to remember the name beta as the beta bend connects two antiparallel beta strands. The term beta comes from the fact that it is a member of a class of turns named with Greek letters, including alpha-, gamma-, delta-, pi-, and beta- turns.
They are almost always on the surface and usually contain 4 amino acids. However, several types of beta turns and different ways to classify them exist. One involves the number of residues (n) between the two hydrogen-bonded residues.
n=2: These contain four amino acids. Amino acids 1 and 4 form hydrogen bonds with n=2 amino acids in between. Another way to describe them is that the hydrogen bond between residues 1 and 4 is between the backbone carbonyl O of the ith amino acid and the amide H of the ith+3 aa (three amino acids away), so the structure contains 4 amino acids (ith, ith+1, ith+2, and ith+3). There are two common types:
- Type I: phi 2 = -60, psi 2 =-30; phi 3 = -90, psi 3 = 0; The first amino acid in the actual turn (ith + 1) is in a left-handed alpha helix conformation. Glycine, asparagine or aspartate are stable at this position since glycine is small and the side chains of Asp and Asn can form hydrogen bonds to the main chain. Glycine is usually found in the second position of the actual turn (ith + 2).
- Type II: phi 2 = -60, psi 2=120; phi 3 = 90, psi 3 = 0; The first residue of the actual turn is typically Gly while the second often is a polar amino acid such as Ser and Thr
Here are 2D line drawings showing Type I and Type II beta turns. Figure 4.2.21

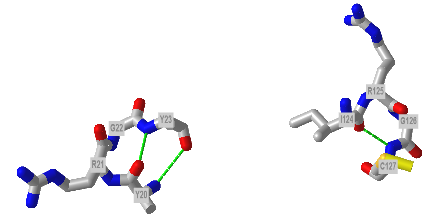
The figure below shows Type I (left) and Type 2 (right) from human egg white lysozyme. Figure 4.2.22
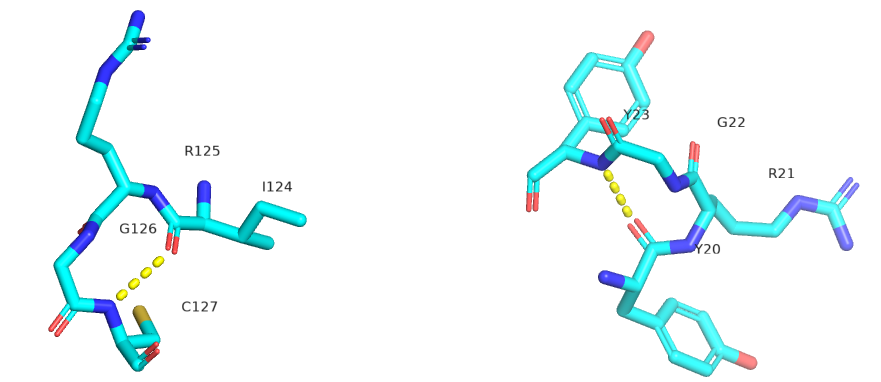
Figure 4.2.23 shows an interactive iCn3D model showing two reverse turns in hen egg white lysozyme (1dpx). Notice the reverse turn's tightness and proline and glycine's presence.
n=3: These contain five amino acids. Amino acids 1 and 5 form hydrogen bonds with n=3 amino acids between them. Another way to describe them is that the hydrogen bond between residues 1 and 4 is between the backbone carbonyl O of the ith amino acid and the amide H of the ith+4 aa (four amino acids away).
Shifting back to the Greek letter naming system, the gamma turn, the second most common turn, has just three total residues (ith, ith+1, and ith+2) with the hydrogen bond between the backbone carbonyl of the ith amino acid and the backbone amide H of the ith+2 amino acid.
An ω-loop is a catch-all term for a longer, extended, or irregular loop without fixed internal hydrogen bonding. Turns are sometimes found within flexible linkers or loops connecting protein domains. Linker sequences vary in length and are typically rich in polar uncharged amino acids. Flexible linkers allow connecting domains to freely twist and rotate to recruit their binding partners via protein domain dynamics.
Mostly, modeling programs display the linear sequence of a protein, and, in addition, a linear cartoon rendering shows the alpha structure as squiggles or helices and the beta structure as yellow arrows. and connecting amino acids between secondary structures as lines. The figure below shows a 1D connectivity diagram for part of the protein alpha-lactalbumin (1a4v). Figure 4.2.24

Amino Acid Propensities for Secondary Structures
Finally, what types of amino acids are most likely found in different secondary structures? Some rationales for the "propensity" for secondary structure are shown in the figure below. Figure 4.2.25
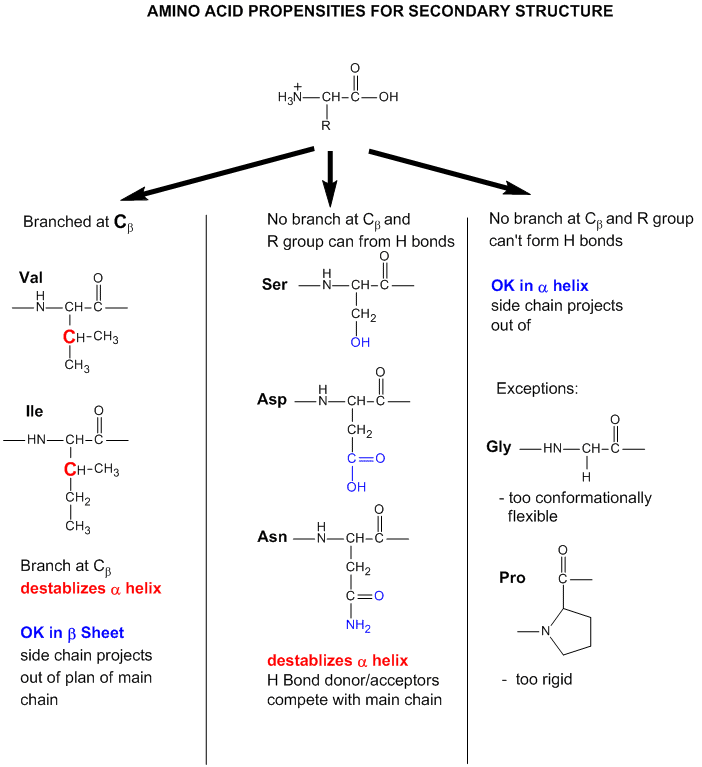
Chou-Fasman derived a list of propensities for each amino acid for secondary structure, based on their occurrence in determined protein structures.. These values are shown in Figure 4.2.26 below.

Figure 4.2.26: Chou-Fasman propensity values. Higher + values represent higher propensities. Jain, Vikas & Tu, Raymond. (2011). Coupled Folding and Specific Binding: Fishing for Amphiphilicity. International journal of molecular sciences. 12. 865-89. 10.3390/ijms12031431. Creative Commons Attribution 3.0 Unported
Summary
This chapter explores the critical elements of protein secondary structure, emphasizing how the repetitive arrangement of backbone hydrogen bonds defines key structural motifs and influences overall protein conformation. The discussion begins with an in-depth look at helical structures, including the most common alpha helix, as well as the less prevalent 3₁₀ and pi helices. These helices are characterized by their specific geometric parameters—such as the number of residues per turn, pitch, and characteristic phi/psi angles—and by the way their side chains project outward, often creating amphipathic surfaces or contributing to membrane-spanning segments.
Next, the chapter examines beta structures, distinguishing between parallel and antiparallel beta sheets. It explains how beta strands, which are more extended than helices, arrange themselves into pleated sheets via inter-strand hydrogen bonds. The orientation of side chains—alternating above and below the sheet plane—and the differences in hydrogen bond geometry between parallel and antiparallel arrangements are discussed, highlighting factors that affect stability and the overall twist of the sheet.
The chapter also covers the connectors that join these regular secondary structures, such as reverse turns, hairpins, loops, and flexible linkers. These regions, often rich in specific amino acids like glycine and proline, allow the polypeptide chain to reverse direction and enable the formation of compact, globular tertiary structures.
To rationalize the conformational preferences of protein backbones, the text introduces the concept of dihedral angles—phi (φ) and psi (ψ)—and describes how they are restricted by the planar nature of the peptide bond, as revealed by resonance stabilization. Ramachandran plots are employed as a powerful tool to visualize the allowed and disallowed regions of backbone dihedral space, correlating these with common secondary structure elements.
Finally, the chapter reviews amino acid propensities for forming various secondary structures, using approaches like the Chou-Fasman analysis to explain why certain residues favor alpha helices, beta sheets, or turns. This integrated overview provides a foundation for understanding how the recurring patterns of hydrogen bonding and the intrinsic properties of amino acids combine to dictate the folding and functional architecture of proteins.
Overall, the chapter lays the groundwork for a deeper exploration of protein tertiary and quaternary structure by elucidating the fundamental principles that govern the formation and stability of secondary structure elements in proteins.
OpenStax, Proteins. OpenStax CNX. Sep 30, 2016 http://cnx.org/contents/bf17f4df-605c-4388-88c2-25b0f000b0ed@2.
File:Chirality with hands.jpg. (2017, September 16). Wikimedia Commons, the free media repository. Retrieved 17:34, July 10, 2019 from commons.wikimedia.org/w/index.php?title=File:Chirality_with_hands.jpg&oldid=258750003.
Wikipedia contributors. (2019, July 6). Zwitterion. In Wikipedia, The Free Encyclopedia. Retrieved 21:48, July 10, 2019, from en.Wikipedia.org/w/index.php?title=Zwitterion&oldid=905089721
Wikipedia contributors. (2019, July 8). Absolute configuration. In Wikipedia, The Free Encyclopedia. Retrieved 15:28, July 14, 2019, from en.Wikipedia.org/w/index.php?title=Absolute_configuration&oldid=905412423
Structural Biochemistry/Enzyme/Active Site. (2019, July 1). Wikibooks, The Free Textbook Project. Retrieved 16:55, July 16, 2019 from en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Enzyme/Active_Site&oldid=3555410.
Structural Biochemistry/Proteins. (2019, March 24). Wikibooks, The Free Textbook Project. Retrieved 19:16, July 18, 2019 from en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Proteins&oldid=3529061.
Fujiwara, K., Toda, H., and Ikeguchi, M. (2012) Dependence of a α-helical and β-sheet amino acid propensities on teh overall protein fold type. BMC Structural Biology 12:18. Available at: https://bmcstructbiol.biomedcentral.com/track/pdf/10.1186/1472-6807-12-18
Wikipedia contributors. (2019, July 16). Keratin. In Wikipedia, The Free Encyclopedia. Retrieved 17:50, July 19, 2019, from en.Wikipedia.org/w/index.php?title=Keratin&oldid=906578340
Wikipedia contributors. (2019, July 13). Alpha-keratin. In Wikipedia, The Free Encyclopedia. Retrieved 18:17, July 19, 2019, from en.Wikipedia.org/w/index.php?title=Alpha-keratin&oldid=906117410
Open Learning Initiative. (2019) Integumentary Levels of Organization. Carnegie Mellon University. In Anatomy & Physiology. Available at: https://oli.cmu.edu/jcourse/webui/syllabus/module.do?context=4348901580020ca6010f804da8baf7ba.
Wikipedia contributors. (2019, July 16). Collagen. In Wikipedia, The Free Encyclopedia. Retrieved 03:42, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Collagen&oldid=906509954
Wikipedia contributors. (2019, July 2). Rossmann fold. In Wikipedia, The Free Encyclopedia. Retrieved 16:01, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Rossmann_fold&oldid=904468788
Wikipedia contributors. (2019, May 30). TIM barrel. In Wikipedia, The Free Encyclopedia. Retrieved 16:46, July 20, 2019, from en.Wikipedia.org/w/index.php?title=TIM_barrel&oldid=899459569
Wikipedia contributors. (2019, July 16). Protein folding. In Wikipedia, The Free Encyclopedia. Retrieved 18:30, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Protein_folding&oldid=906604145
Wikipedia contributors. (2019, June 11). Globular protein. In Wikipedia, The Free Encyclopedia. Retrieved 18:49, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Globular_protein&oldid=901360467
Wikipedia contributors. (2019, July 11). Intrinsically disordered proteins. In Wikipedia, The Free Encyclopedia. Retrieved 19:52, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Intrinsically_disordered_proteins&oldid=905782287