
1.11: Recombinant DNA Technology
- Page ID
- 73676

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Understand the importance of recombinant DNA technology.
- Learn isolation of DNA and its separation on an agarose gel.
- Understand restriction and ligase enzymes and their application in gene cloning.
- Understand vectors and their application in gene cloning and expression.
- Understand polymerase chain termination reaction (PCR).
Introduction
Recombinant DNA (rDNA) technology has resulted in breakthroughs in crop and animal biotechnology. The power of rDNA technology comes from our ability to study and modify gene function by manipulating genes and transform them into cells of plant and animals. To arrive at this several tools of molecular biology are used including, DNA isolation and analysis, molecular cloning, quantification of gene expression, determination of gene copy number, transformation of the appropriate host for replication or transfer into crop plants and analyses of transgenic plants.
Definition and background
Recombinant rDNA technology involves procedures for analyzing or combining DNA fragments from one or several organisms (Figure 1) including the introduction of the rDNA molecule into a cell for its replication, or integration into the genome of the target cell.
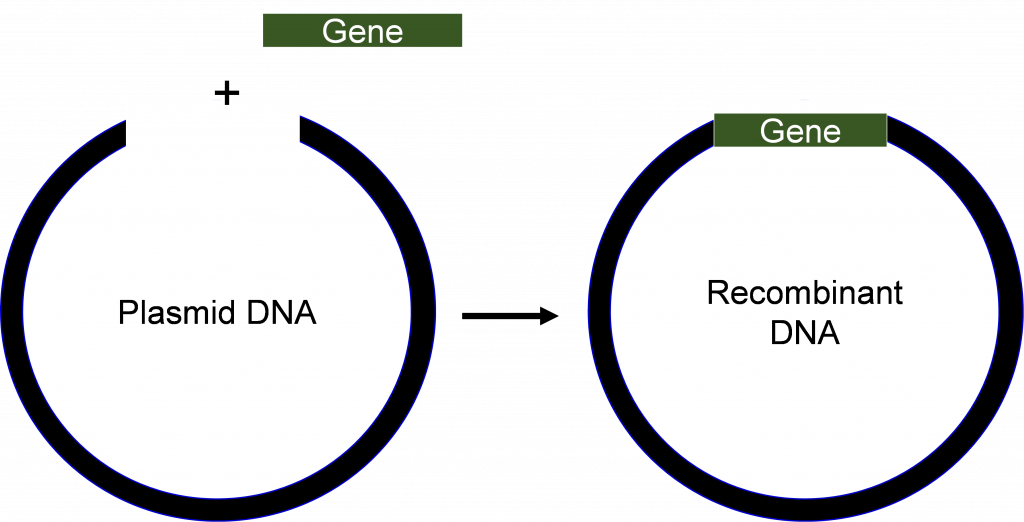
Advances in molecular biology in the early 1970s, including the success in creating, and transferring DNA molecules into cells, revolutionized both science and industry. The first genetically modified organisms were bacteria that made simple proteins of pharmaceutical interest, for example, insulin. As the technologies improved, other organisms including plants became amenable for improvement by rDNA technology. Table 1 provides important milestones in the development and application of rDNA technology.
|
Event |
Year
Mendel’s experiments published
1866
DNA discovered in cell
1869
Mutation of genes by x-rays
1927
One gene-one enzyme hypothesis
1941
DNA is identified as the genetic material
1944
Structure of DNA determined
1953
Ribosomes synthesize protein
1954
Function of mRNA proposed
1961
Genetic code determined
1961-64
Isolation of a restriction enzyme
1970
Recombinant DNA techniques developed
Early 1970s
Isolation of a single copy gene from higher eukaryote
1977
Rapid method of DNA sequencing developed
1977
Plant transformation
1983
Field testing of transformed plants
ca. 1986
Release of engineered plants to general public in the US
1995-96
Transformation of cells with rDNA produces organisms called bioengineered or genetically modified organisms (GMOs). The GMOs contain new traits from another organism. The first GMOs were Escherichia coli cells that were transformed with genes from human to produce various proteins for pharmaceutical purposes.
Isolation of DNA, restriction digestions and its separation on an agarose gel:
Preparation of DNA
For recombinant DNA procedures to work, a pure DNA sample must be obtained. The challenge is that plant and animal cells produce numerous other compounds that often act as contaminants and may inhibit cloning or sequencing of the DNA. Also, tissues and organs from the same plant, or different plants often contain different composition of metabolites, for example, proteins, lipids, and carbohydrates. These compounds must be separated from the DNA during isolation. To achieve this, scientists take advantage of the chemical and physical properties of different molecules inside the cell. For example, DNA is negatively charged, making it soluble in aqueous solution. However, the polar sugar phosphate groups of the DNA are repelled by non-polar solutions. Therefore, the final step in many DNA purifications protocols involves precipitation using alcohol. Other compounds, for example, proteins can also be easily separated from DNA by altering the concentration of salt in the extraction buffer.
Digestion of DNA with restriction endonucleases
Restriction endonucleases are a group of enzymes derived (primarily) from bacteria. Although there are several different types of restriction enzymes, those most useful for rDNA technology recognize specific short sequences in DNA and cleave the DNA at that site to produce cohesive (sticky) or blunt-ended fragments (Figure 2).

More than 500 different restriction enzymes have been identified and can be purchased commercially. Thus, one may ask the question, how often does a restriction enzyme cut within a genomic sequence? It is not possible to give an exact answer for this question. However, let us assume that there are 4 bases on any strand of DNA. This means the probability of detecting an A (adenine) at a particular location is 1/4. Now, since most restriction enzymes recognize specific sequences of 6 bases long, the probability of finding such a site is (1/4)6 = 1 site in every 4,096 base pairs (bp). Assuming you have isolated genomic DNA from maize, and you want to digest it with a restriction enzyme that cuts every 4,096 (4,100) bp, how many fragments will you obtain? To answer this question, you need to have an idea of the size of the genome of the plant you are working with. The approximate size of the maize genome is 2,500,000,000 bp. Thus, using an enzyme that cuts every 4,100 nucleotides one would expect to obtain 2,500,000,000 bp/4,100 bp, approximately 610, 000 fragments. Note that nucleotide distribution is not always random and thus frequency may be different for given DNA. Also, methylation of specific bases in genomic DNA can prevent cleavage at some site.
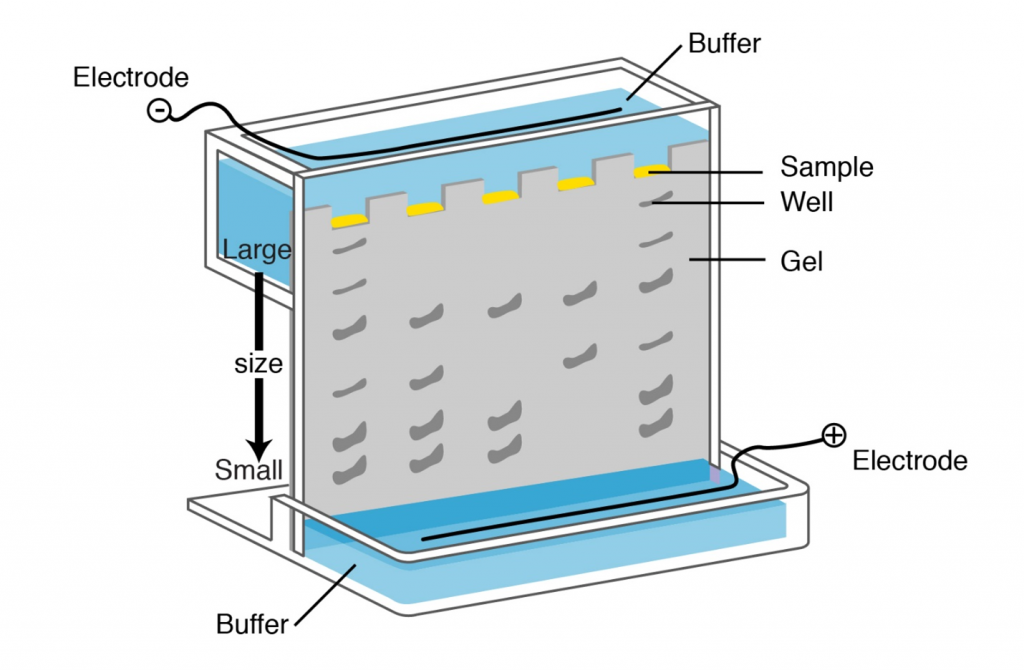
Separation of digested genomic DNA on agarose gel
The only physical features of nucleic acid fragments that are routinely used for their characterization are their size and nucleotide sequence. Molecular weight of DNA is most conveniently evaluated by electrophoresis in agarose gels. Agarose forms a gel by hydrogen bonding when cooled from the melted state. This gel, interwoven network of agarose chains, interferes with the movement of DNA through the gel (see Lesson on PCR and Gel Electrophoresis). Pore size, which affects rate of movement of DNA fragments of a given size, depends on the concentration of agarose. The gel is submerged in electrolyte solution; sample is loaded into wells on one end and current is applied to facilitate movement of DNA fragments. Since DNA is negatively charged it will migrate in the electrical field. Fragments separate according to size. The distance of the migration in each time is proportional to 1/log MW. Following gel electrophoresis DNA can be visualized by staining with ethidium bromide (see Lesson on PCR and Gel Electrophoresis) or other DNA stains.
Tools for gene (DNA) cloning
Polymerase Chain Reaction (PCR)
Polymerase chain reaction (PCR) is a method by which millions of copies of a DNA fragment are produced in a test tube in a matter of minutes or a few hours. The basic steps in PCR reaction were discussed in the Lesson on PCR and Gel Electrophoresis.
Once the sequence of a particular gene is known, it becomes possible to use PCR to isolate that gene from any DNA sample. Recall in Lesson on Gene Transcription you learned that at the genomic level a gene is made up of regulatory sequences, coding, and non-coding sequences. Thus, if the goal is to use PCR to isolate both regulatory and non-regulatory sequences of a gene, the approach would be to use DNA as the starting material.
In cloning genes by PCR, restriction enzyme sites are added at the 5′-end of the primers to facilitate cloning of PCR fragments. A few additional nucleotides (~6 nucleotides) added at the 5′-end of the restriction sites to facilitate restriction digestion of PCR products prior to cloning in a plasmid vector. Alternatively, PCR products may be cloned directly into a T-vector without restriction digestions in E. coli (read more about Promega cloning vector systems). After cloning into E. coli, the fragment is analyzed by sequencing and then sub-cloned into suitable vectors for expression studies.
Like all other biochemical processes, DNA synthesis by PCR is not a perfect process, and occasionally the polymerase enzyme will add an incorrect base to the growing DNA strand. In the context of DNA replication in a cell, the errors are corrected by the DNA polymerase, this is called “proofreading”. Commercially available polymerases may or may not have proofreading capability.
Another important consideration in PCR analysis is contamination.
Minor contamination of the starting material can have serious consequences. Recall that minute amounts of starting DNA can be amplified to millions of copies through PCR. If one inadvertently (or carelessly) mixed DNA from two different sources, the results will be confounding making it impossible to distinguish lines and may cost a laboratory time and money. Ensure that proper procedures are followed in preparing PCR assays. One common source of contamination in plant biology is the products from previous amplification processes. A completed PCR reaction will contain millions of copies of amplified fragments so that even a minute droplet or aerosol from a pipette tip will contain an enormous number of amplifiable molecules. It is always essential to run negative controls which will reveal the presence of contaminating DNA in your PCR assays.
Cloning vector definition and requirements
A cloning vector is a specialized DNA sequence that can enter a living cell and provide means for detection of its presence to a researcher by conferring a selectable property on the host cell (e.g., resistance to antibiotics), and possess means for self-replication. A vector must also possess easily distinguishable physical traits, such as size, or shape, to allow purification away from the host cell’s genome.
Ligase enzyme and gene cloning
Cutting and joining together of vector and DNA fragments from different origins results in rDNA. Recall that restriction endonucleases are used to cut DNA. To join DNA molecules together, an enzyme called DNA ligase is used. The enzyme DNA ligase is used to seal together restriction fragments by forming new phosphodiester bonds. The ligated vector and DNA fragment can now be transformed into a host cell for replication and expression.
The transformation of E. coli takes several steps. First, a gene of interest is inserted into a plasmid that contains a selectable marker usually encoding for resistance to an antibiotic. Second, the plasmid construct containing the gene of interest is transformed into bacterial cells by briefly exposing the mixture of ligated plasmid-DNA fragment (rDNA molecule) and bacterial cells to cold (0oC) and heat (37-42oC). The next step is to grow the transformed cells on selection media containing an antibiotic. Only the cells that have been transformed with the plasmid containing the gene of interest and the marker for resistance to the antibiotic will survive. In addition to using an antibiotic, plasmid vector systems that contain the lacZ gene encoding β-galactosidase allow for easier selection of positive colonies that may harbor the rDNA molecule of interest.
Types of cloning vectors
An example of a cloning vector is a plasmid (Figure 4), defined as an autonomously replicating extra chromosomal circular DNA which is faithfully passed on to progeny. Plasmids are double stranded circular DNA and range in size from about 1 kb – 200 kb. The most useful for cloning are 2 – 10 kb because smaller plasmids are easier to manipulate and usually produce higher copy numbers when grown in bacterial host cells.
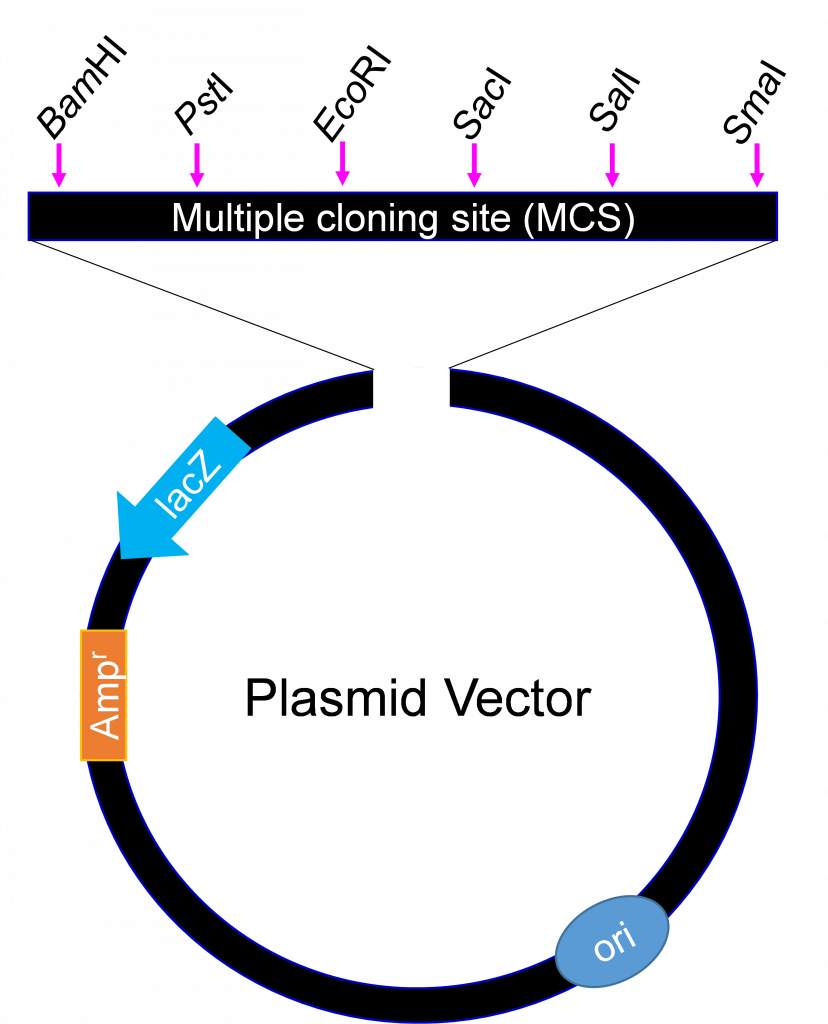
Generally, no more than 10 kb is cloned into plasmids. For cloning large DNA fragments with high efficiency, a vector called bacteriophage lambda is used. Large chromosomal DNA fragments close to 23 kb are stable when introduced into a lambda phage vector.
mRNA as starting material for gene cloning
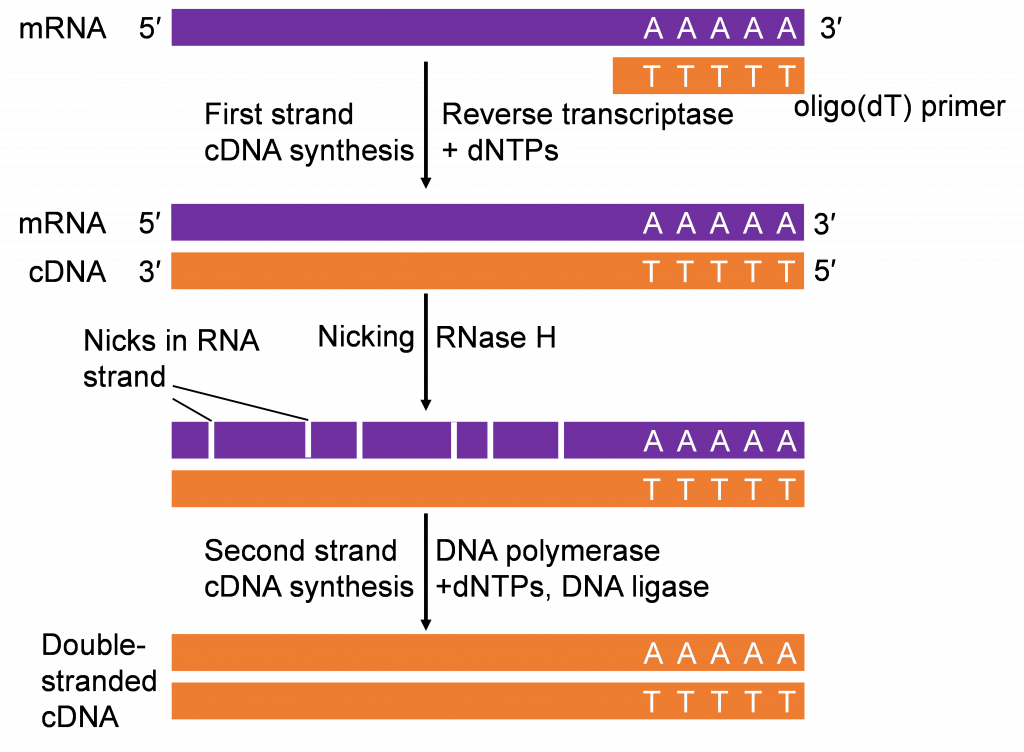
Recall in the Lesson on Transcription you learned about synthesis of RNA from DNA through the process of transcription. Transcription is an important step in gene expression. The mRNA produced can be isolated and “copied” back to DNA by a process called reverse-transcription (Figure 5). The first step of reverse transcription mimics a strategy used by retroviruses (e.g., HIV) that have RNA genomes. As part of their gene-transmission package, retroviruses also contain an enzyme called RNA-dependent DNA Polymerases, commonly referred to as reverse transcriptase. After infecting a host cell the retrovirus uses its reverse transcriptase to copy its single stranded RNA genome into a strand of complementary DNA (cDNA). The reverse transcriptase then synthesizes the second DNA strand from the first strand to make a double stranded-DNA copy which integrates into the host genome.
If only a gene’s coding sequence is required, isolating the gene from cDNAs would be the strategy. It is important that cDNAs are synthesized from tissues expressing the gene of interest. Thus, prior knowledge of where the gene is functional is important in constructing the cDNA molecules for cloning the gene of interest.
The cDNAs produced in vitro (Figure 4) can be used for PCR analysis, similar to chromosomal DNA. The combination of reverse-transcription and PCR (RT-PCR) is a valuable tool in gene cloning and quantification of mRNA.
Recombinant DNA technology has contributed significantly to development of agricultural biotechnology. Transformation of cells with rDNA produces organisms called bioengineered or genetically modified organisms. The tools for plant rDNA technology include, vectors, restriction enzyme, ligation enzymes, bacterial hosts, methods to isolate and multiply nucleic acids, methods to quantify nucleic acids, Agrobacterium as a vector to insert foreign DNA into plants.
Activity 1
Below is a hypothetical DNA fragment containing restriction sites for EcoRI and BamHI.
- How many fragments will be produced after cutting the DNA with BamHI? What size will these fragments be in bp?
- How many fragments will be produced after cutting the DNA with both BamHI and EcoRI at the same time? What size will these fragments be in bp?
- How many fragments will be produced after cutting the DNA with EcoRI? What size will these fragments be in bp?
Activity 2
- You need to clone an EcoRI fragment in the EcoRI site of an expression plasmid vector in E coli. The fragment is 1,809 bp long. There is a BamHI site in the fragment at 1,204 bp. In the plasmid vector, there is a single BamHI site at the promoter region for expression of the gene in E. coli. The BamHI site is 100 bp upstream of EcoRI cloning site.
Following cloning the EcoRI fragment you will get two classes of clones, only one of which will produce the protein. Describe the approach you will take to identify the correct class of clones for expression of the gene in E. coli.