
3.6: cDNA and Genomic Libraries
- Page ID
- 18138

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Isolation of corresponding genetic information
Instead of synthesizing a desired gene, can we used the amino acid information to directly isolate the corresponding genetic information?
- There are two general sources of genetic information:
- Genomic DNA
- mRNA
If we are considering genomic DNA from eukaryotes, then there are a couple of things to consider:
- The coding region for a gene of interest may be interrupted by one or more intron regions, and thus the complete coding region could be quite long.
- To a first approximation, it does not matter which tissue we use to isolate the genomic information, i.e. the genomic content is the same in all tissues.
If we are considering mRNA from eukaryotes, we may realize the following advantages:
- Introns will be spliced out and the mRNA will contain a contiguous coding region.
- Tissue specific expression of the protein of interest may allow us to isolate appropriate mRNA at enhanced levels, i.e. in tissues where the protein is expressed the mRNA levels are considerably higher than the corresponding genomic levels (there are many more molecules of mRNA than copies of the gene).
Libraries
A "library" is a convenient storage mechanism of genetic information.
- They are typically either "genomic" or "cDNA" (i.e. mRNA in DNA form) genetic information.
- Deduced genetic sequences from corresponding polypeptide information can be used to identify specific genetic information within a library.
cDNA library construction
The enzyme responsible for this is an RNA dependent DNA polymerase called reverse transcriptase.
- Reverse transcriptases have traditionally been isolated from viruses whose genome is actually in an RNA form and must be converted to duplex DNA.
- These viruses typically carry a functional reverse transcriptase along with their mRNA genetic component when they infect cells.
- One of the most common commercially available reverse transcriptases is Moloney murine leukemia virus (MMLV).
- This RNA dependent DNA polymerase (as will all polymerases) add nucleotides to a nacent polynucleotide in the 5' to 3' direction using RNA as the template. It does not contain any 3'->5' exonuclease (proofreading) activity.
MMLV will use mRNA as a template, but requires a primer (it can extend a DNA primer but cannot synthesize one).
- One of the really neat things about eukaryotic mRNA's is the presence of the 3' poly A tracks.
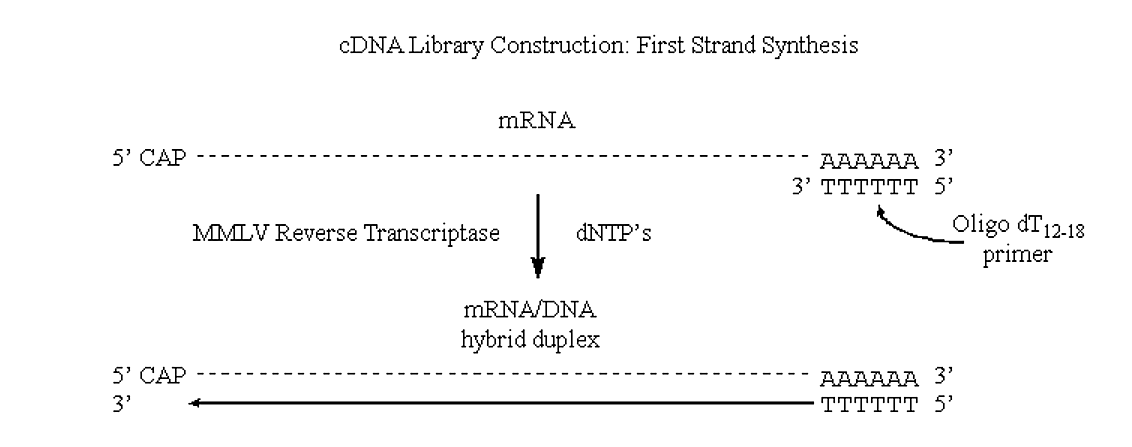.png?revision=1&size=bestfit&width=796&height=314)
If we can introduce "nicks" into the RNA half of this DNA/RNA duplex then the situation would be very similar to that observed in "lagging strand" synthesis of prokaryotic genomic DNA.
- Nicks in the RNA half of the molecule can be introduced via the action of the enzyme RNAse H.
- This enzyme exhibits endonucleolytic cleavage of the RNA moiety of RNA/DNA hybrids, as well as 5'->3' and 3'->5' exoribonuclease activity.
- In other words, it will nick the RNA and then proceed to digest back in both directions:
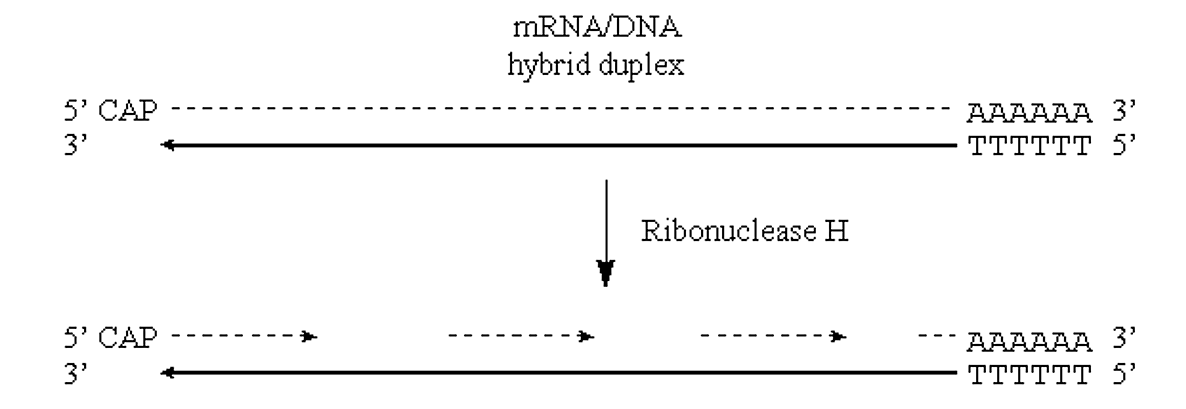.png?revision=1&size=bestfit&width=792&height=273)
- These RNA fragments can now serve as primers for DNA synthesis by E. coli Pol I. This enzyme will also translate the "nicks" to effectively remove the RNA primers:
.png?revision=1&size=bestfit&width=709&height=348)
Figure 3.6.3: DNA synthesis
Insertion of cDNA into plasmid.
To complete our construction of a useful cDNA library we need a way to maintain and propagate our cDNA.
- We can accomplish this by inserting the cDNA into an appropriate plasmid.
- There are two classical ways of accomplishing this feat:
- Homopolymeric tailing
- Linker addition
Homopolymeric tailing
Terminal transferase is an unusual DNA polymerase found only in a type of eukaryotic cell called a prelymphocyte.
- In the presence of a divalent cation the enzyme catalyzes the addition of dNTP's to the 3'-hydroxyl termini of DNA.
- When the nucleotide to be added is a purine, Mg2+ is the cation used.
- When the nucleotide to be added is a pyrimidine, Co2+ is used.
- Depending on the reaction conditions, anywhere from three to several thousand bases will be added.
.png?revision=1&size=bestfit&width=743&height=248)
Figure 3.6.4: Terminal transferase activity
- If we cut our plasmid and also treat it with terminal transferase, except now we add the complementary base to the one we added to our cDNA, we can anneal and ligate the cDNA into the plasmid.
.png?revision=1&size=bestfit&width=408&height=371)
Figure 3.6.5: Ligating cDNA into the plasmid
- The utility of inserting the C-tailed cDNA insert into a G-tailed Pst I site in the vector is as follows:
- The Pst I recognition sequence and cleavage site is
5' C T G C A G 3'
3' G A C G T C 5' - Cleavage of this site by Pst I, followed by G-tailing will produce
5' C T G C A (G)n G 3'
3' G (G)n A C G T C 5'
Linkers
An alternate method to insert cDNA fragments into a library vector is through the addition of "linkers".
- Linkers are short oligonucleotides (~18 to 24 mers) which are typically palindromic and contain a single or repeated restriction endonuclease recognition sequence.
- The palindromic nature allows the linker oligonucleotide to self-hybridize to form a blunt ended duplex.
- If the ends of the cDNA fragments are blunt, then the linker can be ligated to both ends to introduce useful terminal restriction sites.
The steps in linker addition are as follows:
- Treatment of cDNA with S1 nuclease (to remove possible 5' cap mRNA fragment remaining in cDNA duplex
- Convert potential "ragged" ends to blunt by treatment with Pol I (will fill in 5' overhangs and chew back 3' overhangs)
- Methylate cDNA at potential internal Eco RI sites by treatment with Eco RI methylase (plus S-adenosyl methionine)
- Ligate linkers to blunt, methylated cDNA using T4 DNA ligase
- Cut linkers with Eco RI restriction endonuclease
- Remove linker fragments from cDNA fragments by agarose gel electrophoresis
- Ligate cDNA to vector DNA fragment (opened up by Eco RI restriction endonuclease
Editorial Note
This textbook was published in 1998. The Human Genome Project was completed in 2003.
Genomic DNA libraries
Size of some genomes and chromosomes:
|
|
|
| (yeast chromosome 3) |
|
| Escherichia coli (bacterium) genome |
|
| Largest yeast chromosome now mapped |
|
| Entire yeast genome (completed 5/96) |
|
| Smallest human chromosome (Y) |
|
| Largest human chromosome (1) |
|
| Entire human genome |
|
- The human genome contains approximately 50,000 unique genes within 3-4 billion base pairs of DNA, scattered about in 23 pairs of chromosomes.
Fragmentation of genomic DNA for library construction
Restriction endonuclease digestion
- A six-cutter (e.g. Eco RI) will cut on average every 4.1 Kb. Complete digestion of human DNA with this type of enzyme will result in approximately 1 x 106 unique fragments.
- What is the probability of finding a clone within a given library?
The exact probability of having any given DNA sequence in the library can be calculated from the equation
For example, how large a library (i.e. how many clones) would you need in order to have a 99% probability of finding a desired sequence represented in a library created by digestion with a 6-cutter?
Thus, from this type of analysis we can see that we need a technology which will allow us to achieve the following:
- Stable insertion of relatively large DNA fragments into our cloning vector
- High efficiency of insertion and the ability to handle large numbers of clones
- For example, when plating E. coli colonies on a 3" petri plate, the maximum practical density to allow isolation of individual colonies is about 100-200 colonies per plate.
- If we were to try to plate our library of 3.37 x 106 in such a way would need about 22,500 plates.
- Not only that, but such large DNA fragments are not well tolerated in typical E. coli cloning vectors such as pBR322.
Bacteriophage lambda vectors are commonly used for construction of genomic libraries
Bacteriophage l is an E. coli phage with a type of icosahedral phage particle which contains the viral genome:
.png?revision=1&size=bestfit&width=257&height=266)
Figure 3.6.6: Bacteriophase l
- During replication, the phage DNA is produced in a concatameric form, which is cleaved by appropriate endonucleases to allow packaging of a single genome within the phage capsid.
- It was found that internal regions of the phage genome, which were not essential to phage replication, could be removed and replaced with DNA of interest.
- This hybrid DNA could be efficiently packaged, and form an infective phage.
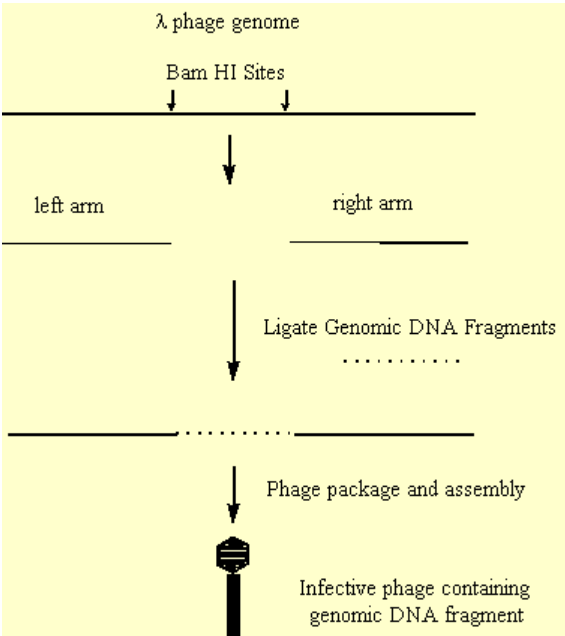.png?revision=1&size=bestfit&width=413&height=465)
Figure 3.6.7: Creation of ineffective phage
The advantages of this type of system vs plasmids like pBR322 are:
- The phage genome is able to package efficiently with DNA inserts as large as 20 Kb.
- Furthermore, the packaged phage are highly infectious and infect E. coli at a much higher efficiency than plasmid transformation methods.
Incomplete Digestion of Genomic DNA will allow identification of sequence overlaps
Complete digestion with an endonuclease will result in a library containing no overlapping fragments:

However, incomplete digestion will result in a library containing overlapping fragments:

- Thus, the sequence information obtained from one clone will allow the isolation of clones containing neighboring (overlapping) sequence information.
- This can allow large contiguous stretches of sequence information to be obtained ("Chromosome Walking").
Probing libraries
Once a library (cDNA or genomic) has been constructed we want to be able to identify clones which contain DNA of interest.
- For example, from protein sequence information we can deduce possible stretches of the corresponding DNA sequence (there will however be ambiguity due to the degeneracy of codons).
- If we can synthesize an oligonucleotide complementary to our DNA sequence of interest we can use it to specifically hybridize to the appropriate clone in our libraray (i.e. to probe our library).
In standard methodologies the oligonucleotide is phosphorylated at the 5' end with radiolabeled g32P-ATP and T4 polynucleotide kinase.
- The probe is then incubated with individual phage plaques which have been fixed onto nitrocellulose and their DNA denatured by treatment with base.
- If the plaque contains complementary DNA to to probe sequence, the probe will hybridize.
- If the nitrocellulose (containing many individual plaques) is exposed to x-ray film, only those plaques with hybridized probe will show up (as a dark spot):
.png?revision=1&size=bestfit&width=323&height=434)
Figure 3.6.8: Radiolabeled plaque
False positives
If we are designing DNA probes from protein sequence information we will have possible ambiguity in our deduced DNA sequence used for the design of the probe.
- Usually 14-24mer oligonucleotides are used as probes, a 14-24mer probe means we need a stretch of 5-8 amino acids in the polypeptide.
- Given the choice, the best amino acid sequences to look for in a polypeptide are those with low codon degeneracy (see above).
- Thus, we would look for a short stretch of polypeptide sequence hopefully containing Met or Trp, and with the remaining amino acids comprising either Phe, Tyr, His, Gln, Asn , Lys, Asp, Glu or Cys.
- Regions including Leu, Arg or Ser are to be avoided (6 codons each).
During oligonucleotide synthesis multiple bases will be incorporated at ambiguous positions.
- Thus our probe will actually be a mixture of oligonucleotides.
- The higher the degeneracy, the greater the posibility of "false positives", i.e. clones which hybridize but are unrelated to the actual sequence we want.
- Positive clones are sequenced and the deduced amino acid sequence is compared to our polypeptide sequence information to identify correct clones.
Antibodies (Immunoglobulins)
If the particular vector, or phage, used to construct a cDNA library contains a promoter region upstream of the insertion site we may be able to screen for desired clones by looking for expression of the protein of interest.
- In this case, we need an assay which is both sensitive (we will not be producing a lot of protein) and specific (we want to minimize any false positives).
- One of the best assays, which is both sensitive and specific, makes use of antibodies.
Antigen, antibody, epitope
One of the defense mechanisms of vertebrates is the ability to distinguish between self and non-self molecules.
- Thus, if a foreign molecule (either from another species or sometimes from another individual within a species) invades a vertebrate organism, the immune system functions to learn to identify that molecule.
- In future invasions by the same molecule, the organism mounts a defense against it by producing specific antibodies which recognize and bind to the foreign antigen.
- When antibodies bind to antigen certain white blood cells (macrophages and monocytes) recognize the invading body as foreign and respond by destroying it.
Antibodies are 'Y' shaped molecules which contain two identical heavy chains, and two identical light chains.
- The stem of the 'Y' comprises the Fc (constant) domain, and the 'arms' of the 'Y' comprise the Fab (variable) domains.
- Antigens bind to the complementarity-determining regions (CDR's) located at the ends of the Fab domains.
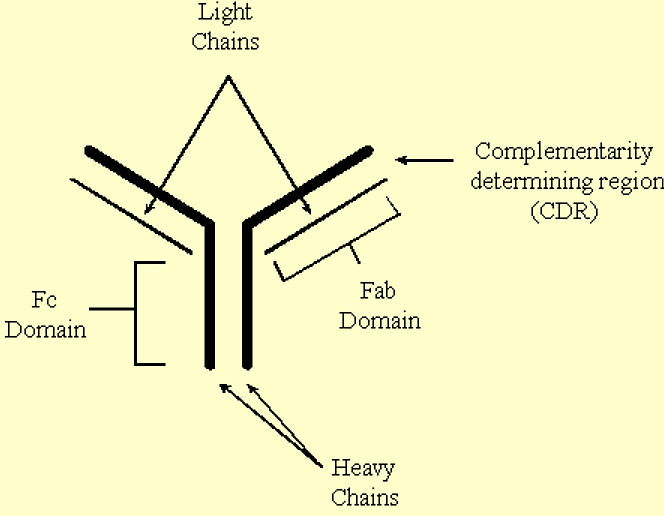.png?revision=1&size=bestfit&width=467&height=363)
Figure 3.6.9: Antibody structure
Antibodies are synthesized by B lymphocytes. Each B lymphocyte is capable of producing a single type of antibody directed against a specific structural determinant, or epitope, on an antigen.
- Thus, an immune response to a protein antigen may result in a population of B lymphocytes each producing antibodies which recognize a different structural determinant of the foreign protein.
- An epitope may be a contiguous region of 5 or 6 amino acids in the foreign polypeptide, or the epitope may comprise a half dozen or so amino acids brought in juxtaposition in the native protein, yet widely spaced in the polypeptide sequence.
- Thus, some antibodies will recognize native and denatured forms of a foreign protein equally well, while other antibodies may only recognize one or the other.
If the protein of interest has been purified it can be used to induce an immune response in a host animal.
- Typical host animals include mouse, chicken, rabbit, goat, sheep, horse and occasionally, human.
- After an initial immunization, followed by one or more booster shots, the B lymphocytes of the host animal may produce antibodies directed against the antigen.
- The antibodies can be be purified from blood samples withdrawn from the animal. Such preparations of antibodies are said to be polyclonal.
- This refers to the fact that the antibodies present are from a collection of different B lymphocytes and thus will recognize a variety of different epitopes on the antigen protein.
- The ability to isolate antibodies from blood samples means that the host animal does not need to be destroyed.
- Of course, the size of the animal determines how much antibodies one can obtain. For example, a rabbit can provide 5 mls of blood every two weeks, a mouse provides significantly less, while a horse can provide quite a bit more.
An antibodiy isolated from a single B lymphocyte cell population is termed monoclonal.
- It recognizes a single epitope on the antigenic protein.
- Antibody producing B lymphocytes can be isolated from the spleen or from lymph nodes. However, they have a finite life spanin culture, i.e. they will undergo a certain number of cell divisions and then die.
- These cells can, however, be fused with immortal (cancerous myeloma) lymphocytes to produce a hybridoma cell.
- Such a cell is immortal like the myeloma, and produces a specific antibody from the B lymphocyte. The ability to grow indefinitely in culture allows the isolation of useful amounts of specific monoclonal antibodies.
Sometimes immunizing with the protein of interest is problematic: appropriate amounts of purified material cannot be produced, or the protein is itself toxic at the dosage level necessary to produce an immune response.
- If partial sequence information is known, then large amounts of polypeptides representing short fragments of the protein, can be synthesized and used to immunize the animal.
- Often these polypeptides are covalently attached to a carrier protein (typically serum albumin) to enhance the antigenic response.
- Antibodies produced against such peptides will recognize only epitopes within the polypeptide. Thus, even polyclonal antibodies would be quite limited in their epitope recognition.
As with radiolabeled oligonucleotides, antibodies can be used to identify library clones which contain a cDNA of interest. This method would of course rely upon a host vector or phage which contains a promoter upstream from the site of insertion of the genomic DNA.
- Antibodies can be used to screen viral plaques or plasmid clonies which have been bound to nitrocellulose.
- Bound antibodies can be identified using radiolabeled protein A (which binds to immunoglobulins) or via a second antibody (which, like protein A, can recognize general immunoglobulins) which has a dye or dye releasing enzyme covalently attached.