
3.5: Protein Sequencing, Peptide Mapping, Synthetic Genes
- Page ID
- 18137

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Historically, some important disease states were identified as being caused by the lack of an important protein, or the presence of a dysfunctional mutated form of a protein.
- For example, diabetes, types of dwarfism and hemophilia were found to be due to deficiencies in insulin, growth hormoneand clotting factor VIII, respectively.
These diseases could be treated by injecting supplemental doses of purified, or partially purified, preparations of these proteins.
- These proteins were isolated from natural materials, e.g. pig (insulin), human cadaver pituitaries (human growth hormone) or blood fractions pooled from normal donors (factor VIII).
- In most cases, even if the protein was found in relatively abundant supply, the cost of production was substantial.
More often than not, interesting bioactive properties were associated with proteins which could be isolated only in minute quantitites(e.g. the blood clot dissolving protein tissue plasminogen activator).
Also, non-human proteins typically elicited an immune response when injected into humans, thus the human form of a protein was the only useful form.
- If the protein were not readily available from blood, or urine, it would prove impractical to obtain adequate starting materialfor production.
- Unfortunately, if the material were derived from human sources, the possibility existed for the spread of human disease (e.g. hepatitis and the AIDS virus).
If the genetic information for these proteins could be isolated, and then transcribed and translated in an easily scaleable biological system, potentially large amounts of protein could be obtained - and hopefully, relatively cheaply.
With the development of "molecular biology", i.e.
- the structure of DNA,
- the elucidation of the genetic code,
- the identification of transcriptional promoters and ribosome binding sites,
- the isolation of restriction endonuclases,
- the identification of the origin of DNA replication
- the development of plasmids with selectable markers, and
- the culturing of E. coli,
the possibility existed in the mid 1970's to put it all together and produce relatively large amounts of any human protein for therapeutic use.
How would you go about the process of producing large amounts of some important human protein? (i.e. protein purification)
The starting point is typically an assay for a functionality of interest. For example, we may have a hemophiliac whose blood does not clot. However, we find that if we take a sample of his blood and add to it a small amount of blood from a "normal" individual, the hemophiliac's blood will now clot. This will be the basis for our assay.
Using this assay, we will fractionate normal blood using various means - chemical precipitation (with ethanol, or ammonium sulfate), and then various liquid chromatography steps, etc.
- Along the way we will follow where our clotting activity is going.
- Hopefully, at some point we will be unable to fractionate it further and will have a pure protein.
Once we have a pure protein we can begin to characterize it with regard to its amino acid sequence. From there we can ultimately get the gene for the protein and express it.
.png?revision=1&size=bestfit&width=520&height=380)
Figure 3.5.1: Protein production
N-terminal peptide sequence analysis
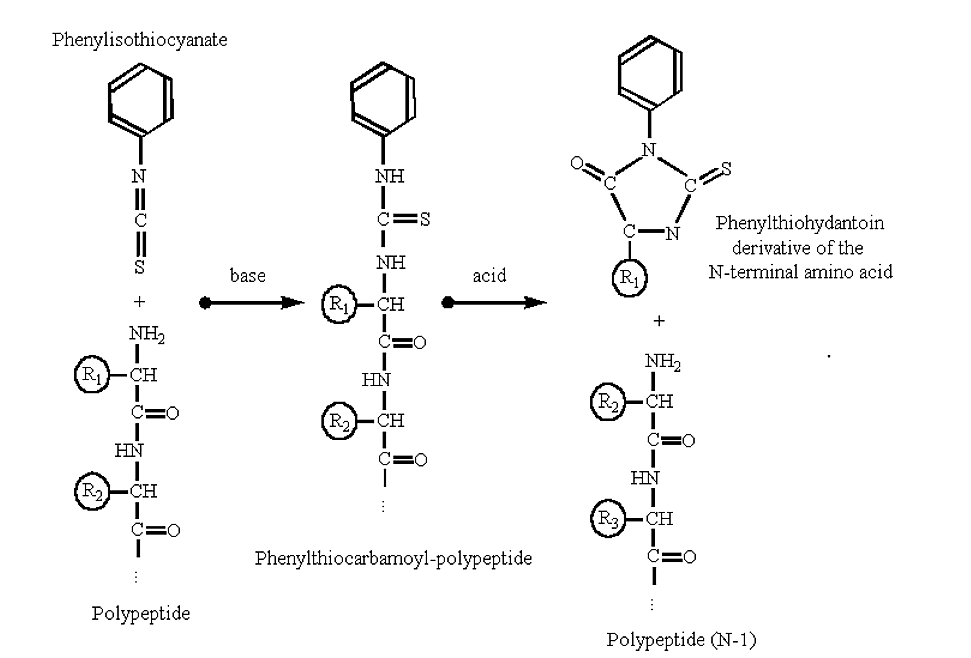
Polypeptides can be sequenced from their amino-terminus by automated procedures based upon the Edman degradation reaction:
.png?revision=1&size=bestfit&width=735&height=497)
Figure 3.5.2: Edman degradation
- Note that with Edman chemistry only the N-terminal residue is attacted and removed, the rest of the polypeptide remains intact after the reaction.
- The new amino terminal group (previously the second amino acid in the polypeptide chain) is now available for another round of reactions. Thus, the method can be automated.
- The amino acid side chain of the phenylthiohydantoin derivative can be identified using liquid chromatography. Modern amino acid sequencers can probably sequence on the order of two to three dozen cycles (amino acids) of a polypeptide.
- Note that the reaction requires a free amino group on the N-terminal of the protein. If the amino-terminal residue is methylated or formylated then the reaction will not proceed (and the polypeptide is said to have a "blocked" N-terminal).
C-terminal peptide sequence analysis
C-terminal peptide sequence analysis is not as well developed as amino terminal analysis.
- The method usually makes use of non-specific carboxypeptidases.
- Carboxypeptidases will sequentially hydrolyze polypeptides from the carboxy-terminus end. The released amino acid can be identified using liquid chromatographic methods, and the remaining polypeptide is available for further reactions.
- Various carboxypeptidases are available, usually they are not entirely non-specific (i.e. they have certain preferences):
|
Name |
Source |
Specificity |
|
Carboxypeptidase A |
Bovine Pancreas |
Aromatics, aliphatics (hydrophobics) |
|
Carboxypeptidase B |
Pig Pancreas |
Arginine, Lysine, Ornithine |
|
Carboxypeptidase P |
Penicillium |
Generally non-specific |
|
Carboxypeptidase Y |
Yeast |
Aromatic, aliphatics |
Sometimes the choice of which carboxypeptidase to use is based upon the expected sequence information. In these types of experiments:
- samples are taken at different time points during the digestion
- free amino acids are separated from polypeptides
- the released amino acids are identified via amino acid analysis (liquid chromatography).
C-terminal analysis is usually only accurately for identification of the last half-dozen residues or so in a polypeptide.
Peptide Mapping
One of the obvious problems with protein sequencing is that even if the N-terminal is not "blocked" only limited sequence infomation can be obtained from an intact polypeptide (i.e. only about two dozen from the N-terminal and half a dozen from the C-terminal).
How can sequence information for the entire polypeptide be obtained?
One method is that of peptide mapping. Peptide mapping makes use of proteolytic cleavages of the polypeptide to produce smaller polypeptides. These smaller polypeptides can then be isoloted from one another and subject to sequence analysis.
How do we order the different sequences which we obtain?
One of the easiest ways is to repeat the experiment, but with a protease with a different specificity, and in this way obtain overlapping sequence information.
|
Name |
Source |
Specificity |
|
Chymotrypsin |
Bovine Pancreas |
Cleavage after Tyr, Phe and Trp; some cleavage after Leu, Met and Ala |
|
Bromelain |
Pineapple |
Cleavage after Lys, Ala and Tyr |
|
Trypsin |
Bovine Pancreas |
Cleavage after Arg, less after Lys |
|
V8 protease |
Staphylococcus aureus |
Cleavage after Glu, less after Asp |
.png?revision=1&size=bestfit&width=627&height=338)
Figure 3.5.3: Overlapping cleavage products
Overlapping sequence information can allow you to align the peptides in the correct order and determine the sequence of the original large polypeptide (i.e. protein).
One problem which can arise deals with Cysteine residues and the nature of any covalent disulfide bridges in the protein.
- Any "peptide" mobilities (on either liquid chromatographic or PAGE analyses) which split into two smaller peptides after treatment with a reducing agent (such as b-ME) indicate the presence of a cysteine mediated disulfide bond.
- Upon sequencing these peptides should each contain a cysteine residue. If each peptide has only one cysteine then the disulfide bond assignment is unambiguous.
.png?revision=1&size=bestfit&width=683&height=401)
Figure 3.5.4: Cysteine residues in cleavage products
Corresponding genetic information
Once we have partial, or complete, peptide sequence information we can begin to identify and isolate the corresponding genetic information. This is the main goal. Once we have the corresponding genetic information it may be possible to produce relatively large amounts of the desired polypeptide.
Back translation
Since we know the genetic code, we can back translate any polypeptide sequence into a corresponding genetic sequence.
- Thus, from the amino acid sequence we could synthesize an artificial gene which would code for the protein of interest.
- Since many amino acids are coded for by more than one codon, there is potential ambiguity with regard to the original exact genetic sequence.
|
Amino Acid |
Number of Codons |
|
Met, Trp |
1 |
|
Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys |
2 |
|
Ile |
3 |
|
Val, Pro, Thr, Ala, Gly |
4 |
|
Leu, Arg, Ser |
6 |
However, making sure we back translate in such a way as to faithfully duplicate the original genetic sequence may not be critical - a correct protein sequence is the overall goal.
In fact, if we are attempting to express the protein in another organism (say expressing a mammalian gene in a bacterial system) we may actually prefer to choose a codon bias appropriate for the expression host organism.
Synthetic genes for small proteins are a reasonable way to proceed; this is one way in which human insulin has been expressed in bacterial systems.
- However, automated synthesis of DNA oligonucleotides is practical for polymer lengths of approximately 60-90 bases or less (about 20-30 amino acids).
- Furthermore, the method of construction of synthetic genes typically calls for overlapping complementary oligonucleotides (to be ligated into a single duplex DNA gene "cassette").
Thus, many oligonucleotides are required for even a single small synthetic gene.
.png?revision=1&size=bestfit&width=587&height=246)
Figure 3.5.5: Synthetic gene construction
One way to improve upon the above method of synthetic gene construction is with a direct PCR approach. This method does not utilize ligase, or even oligonucleotides that butt together. Instead, with this method many (~100) different overlapping oligonucleotides are simultaneously used in a PCR reaction. Their sequence complementarity can be represented as follows:

The entire set of oligonucleotides may not line up to give the entire gene, but that is alright. We will do multiple rounds of PCR with the idea that some complementary oligo's will anneal and be extended and will lead, bit by bit, to construction of a contiguous synthetic gene:
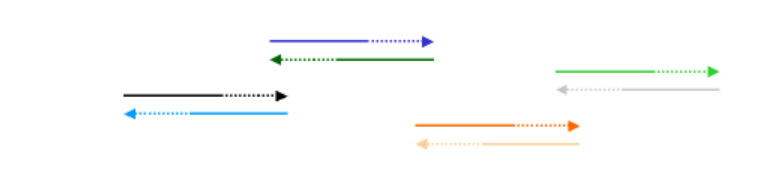
On the next PCR cycle, some of these extended fragments will anneal with others:
.png?revision=1&size=bestfit&width=577&height=160)
These will be extended via the PCR and can go on to anneal with other larger PCR fragments. Eventually, the entire gene will be constructed. However, since the efficiency of construction of the full-length gene is probably not going to be very good, we need to conduct a subsequent PCR experiment to amplify the full-length gene (using outer primers). The principle features of this method are summarized as follows:
- Many (as many as 1-2 hundred) overlapping oligo's are combined in a single PCR reaction
- The oligos are designed to be as long as possible (~100mers) with limited overlap (~20 bases)
- The full-length gene is constructed in an initial (low yield) PCR experiment
- This full length gene is amplified with a subsequent typical PCR experiment using outer primers.