
6.5: Enzyme and Protein Regulation
- Page ID
- 102268


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)(Learning goals written by Claude, Sonnet 4.6, Anthropic)
Post-Translational Modifications and Their Detection
- Describe the major classes of protein post-translational modifications (PTMs) — including phosphorylation of Ser/Thr/Tyr, N- and O-acetylation of the α-amino terminus and Lys ε-amino group, N- and O-linked glycosylation, methylation, ubiquitination, sumoylation, and oxidative modifications — identifying the target amino acid residue, the co-substrate or donor molecule (ATP, acetyl-CoA, SAM, ubiquitin), and the reversibility of each modification, and explain how the amplification from ~20,000 genes to ~200,000 transcripts to potentially ~1,000,000 proteoforms arises from alternative splicing and PTMs.
- Explain the opposing enzymatic systems that regulate reversible PTMs — kinases/phosphatases for phosphorylation, KATs/KDACs and sirtuins for acetylation, and histone methyltransferases/demethylases for methylation — and describe how histone acetylation relaxes chromatin to promote transcription (neutralizing positive Lys charge, reducing histone:DNA interaction) while deacetylation and methylation condense chromatin to suppress transcription, connecting these modifications to epigenetic regulation.
- Explain the ubiquitin–proteasome system — describing how the sequential action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes attaches ubiquitin via its C-terminal Gly 76 to Lys residues on target proteins, how polyubiquitin chains target proteins to the 26S proteasome for degradation, and how monoubiquitination instead regulates protein activity, localization, and protein-protein interactions — and contrast this with SUMO modification, which shares structural homology with ubiquitin but does not target proteins for degradation and instead regulates nuclear transport and protein-protein interactions.
Allosteric Regulation and Zymogen Activation
- Describe how allosteric regulation of enzyme activity can be achieved through diverse mechanisms — small molecule binding inducing hinge-clamping conformational changes (phosphoglycerate dehydrogenase, feedback inhibition by serine), subtle electrostatic rearrangement without large conformational change (chorismate mutase, repositioning of Glu 23 by bound Tyr vs. Trp), phosphorylation-driven T↔R state conversion (glycogen phosphorylase, Ser 14 phosphorylation activating the enzyme), promotion or inhibition of quaternary structure formation (ATP phosphoribosyltransferase, histidine-driven hexamerization closing the active site), and disulfide bond cleavage (botulinum neurotoxin, reduction releasing the catalytic domain from the inhibitory domain) — and distinguish allosteric conformational changes caused by ligand binding at an allosteric site from substrate-induced induced-fit changes at the active site (exemplified by hexokinase).
- Explain the molecular basis of zymogen activation using trypsinogen → trypsin as the model — describing how co-secretion with the pancreatic secretory trypsin inhibitor (PSTI) provides a second safety mechanism against premature activation, how enteropeptidase cleaves the trypsinogen activation peptide (TAP) from the N-terminus in the duodenum to trigger a conformational change revealing the active site, and how active trypsin then activates the cascade of other digestive zymogens (chymotrypsinogen, proelastase, procarboxypeptidase, prolipase) — connecting zymogen activation to the broader principle of irreversible covalent modification as a regulatory strategy.
Isozymes and the COX System: A Pharmacological Case Study
- Compare COX-1 and COX-2 as isozymes — distinguishing their tissue distribution (COX-1 constitutively expressed broadly; COX-2 normally undetectable but induced by inflammatory and mitogenic stimuli), their physiological roles in producing prostacyclin PGI₂ (vasodilation and platelet inhibition, primarily COX-2 in endothelial cells) vs. thromboxane TXA₂ (vasoconstriction and platelet aggregation, primarily COX-1 in platelets), and the structural basis for drug selectivity (the COX-2 side pocket accommodating bulkier coxibs that cannot fit the narrower COX-1 channel, governed by a single Val→Ile substitution at position 523).
- Classify NSAIDs by mechanism — aspirin as an irreversible inhibitor (covalent acetylation of Ser 530), non-selective competitive inhibitors blocking the arachidonic acid channel via interactions with Arg 120 and Tyr 355 (ibuprofen, naproxen), and selective COX-2 inhibitors (coxibs, e.g., celecoxib) exploiting the COX-2 side pocket — and explain the cardiovascular risk of coxibs using the thromboxane/prostacyclin imbalance hypothesis: selective COX-2 inhibition reduces vasodilatory PGI₂ without reducing platelet TXA₂, tipping the balance toward vasoconstriction and thrombosis and explaining the increased myocardial infarction and stroke risk that led to rofecoxib's market withdrawal.
Many different mechanisms control protein activity within a cell. The primary sequence of a protein is a main determinant of protein folding and final conformation, as well as biochemical activity, stability, and half-life. However, at any given moment, the proteome, the full complement of proteins within a cell, is up to two or three orders of magnitude more complex than the encoding genomes would predict. This chapter will provide an overview of the major mechanisms biological systems use to regulate protein function after synthesis. Note that these mechanisms seldom work in isolation. Multiple levels of protein control function at any given time and in response to many different environmental cues and signals. We will discuss many ways to regulate protein activity. At the end, we will discuss regulation through the use of different isozymes of an enzyme, which are variants arising from differential splicing of a gene or from slightly different genes that arose from a common precursor gene. We will focus on cyclooxygenases, the target of so many medicinal drugs.
Post-translational modifications (PTMs)
The human genome contains approximately 20,000 genes. When analyzing the transcriptome, it becomes apparent that the genome becomes amplified by the wide array of splice variants that can occur when primary RNA transcripts are spliced to form mature messenger RNAs. There are about 200,000 expressed transcripts, of which about 146,00 encode proteins, with about 7.4 transcripts per gene. A summary of human gene transcripts is shown in Table \(\PageIndex{1}\) below.
| Type |
GTEx dataset |
Expressed all genes |
Expressed protein-coding genes |
|---|---|---|---|
|
Transcripts per Gene |
3.42 |
3.63 |
7.43 |
|
Gene counts |
58,219 |
53,539 |
19,591 |
|
Transcript counts |
199,324 |
194,146 |
145,571 |
Table \(\PageIndex{1}\): Numbers of transcript and gene in the GTEx dataset. Tung, KF., Pan, CY., Chen, CH. et al. Top-ranked expressed gene transcripts of human protein-coding genes investigated with GTEx dataset. Sci Rep 10, 16245 (2020). https://doi.org/10.1038/s41598-020-73081-5. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
The number of distinct primary protein structures (proteoforms) from 20,000 human genes is further amplified by posttranslational modifications (PTMs), which produce up to a million different variants. PTMs are present in both eukaryotes and prokaryotes. Still, they are more common in eukaryotic cells, where about 5% of the genome encodes enzymes that modify proteins posttranslationally. We discussed the chemical modification of specific amino acids in Chapter 3.1: Amino Acids and Peptides.
Protein PTM results from the enzymatic or nonenzymatic attachment of specific chemical groups to amino acid side chains. Such modifications occur either following protein translation or concomitant with translation. PTM influences both protein structure and physiological and cellular functions. Examples of enzymatic PTMs include phosphorylation, glycosylation, acetylation, methylation, sumoylation, palmitoylation, biotinylation, ubiquitinylation, nitration, chlorination, and oxidation/reduction. Nonenzymatic PTMs include glycation, nitrosylation, oxidation/reduction, acetylation, and succinylation. Some rare and unconventional PTMs, such as glypiation, neddylation, siderophorylation, AMPylation, and cholesteroylation, also influence protein structure and function. Note that many of these modifications are not made in isolation. It is common for proteins to undergo multiple types of modifications, which can vary depending on tissue type and environmental conditions. The major PTMs in eukaryotes, their target amino acid residue(s), and the types of enzyme(s) or protein(s) involved are shown in Table \(\PageIndex{2}\) below.

Table \(\PageIndex{2}\): Common Protein Post-Translational Modifications, Their Target Amino Acid Residues, and the Enzyme(s) or Proteins Involved. Santos, A.L, and Lindner, A.B. (2017) Oxidatve Medicine and Longevity, Article ID: 5716409
PTMs can exert their effects through many structural changes, including opening or closing the active site, altering the conformation and electrostatic properties of binding sites, changing the chain's flexibility, increasing or decreasing intrinsically disordered regions, altering protein-protein interactions, etc. These effects can also arise from the binding of small molecules.
Methods to Detect Protein Posttranslational Modifications
Specific amino acid residues undergo PTMs depending on the reaction chemistry and the sequence specificity of the enzyme involved. Initially, PTMs were detected using various analytical methods, including radiolabeling of proteins, thin-layer chromatography, column chromatography, and/or polyacrylamide gel electrophoresis. Other methods have since been developed, such as Edman degradation for protein sequencing and Western blotting with protein-specific antibodies. Antibody-based detection methods and mass spectrometry-based proteomic analysis are the predominant approaches for detecting and analyzing PTMs. However, mass spectrometry is the only available tool for global or large-scale PTM analysis.
Antibody-based methods mostly rely on antibodies that specifically recognize a modified amino acid residue within a protein or peptide. Such antibodies can be polyclonal or monoclonal and are developed against either the modified peptide/protein or against the modified amino acid. Moreover, antibody-based detection and quantification of PTMs on protein/peptide samples can be performed by chemiluminescence-based Western blotting and absorbance/fluorescence-based ELISA. However, PTM detection depends entirely on the antibody's recognition site. If the antibody detects only the modified amino acid, additional analysis—for instance, protein/peptide isolation and sequencing—should be performed to detect the sequence context of the modification. However, suppose the antibody detects the PTM within a specific sequence context. In that case, the presence of PTM at other sites will remain undetected (ie, the antibody will be specific for only that single modification).
Mass spectrometric detection of specific PTMs is based on mass changes. The mass spectrometer detects a change in mass to identify the presence of a PTM in a peptide sample. Using tandem mass spectrometry, the specific site of a PTM can be identified by subsequent fragmentation and sequencing of the relevant peptide. Yet, technical challenges hamper MS-based investigation of biologically important PTMs, such as ADP-ribosylation, one of the key signaling molecules that regulate DNA repair, a critical process in maintaining genome stability that is compromised in cancer and aging.
Data increasingly implicate PTMs in aging and/or under pathological conditions, as well as in the cell's normal functioning. PTMs are increasingly studied for their role in health and disease. For example, the precise and accurate measurement of distinct PTM-containing moieties offers potential biomarker utility to aid early diagnosis, prognosis, monitoring response to therapy, and decisions regarding inclusion in clinical trials as new medicines are developed. However, technical difficulties limit these studies, leaving many unanswered questions. Identifying unknown/unexpected PTMs by proteomic data reanalysis is an emerging subfield of proteomics, recently boosted by the increased availability of raw data shared in public repositories. Notably, sampling the proteome in a given organism or cell provides only a snapshot of a highly dynamic process, complicating the analytical problem and ultimately arguing for time-resolved inventories. Thus, while many tools are currently available for studying PTMs, new methods are needed to further advance the study of these modifications.
Examples of PTMs
Protein PTMs involve the covalent addition of some chemical groups by enzymatic catalysis. Typically, an electrophilic fragment of a co-substrate is added to an electron-rich protein side chain, which acts as a nucleophile in the transfer. Common covalent protein PTMs include phosphorylation, acylation, methylation, sumoylation, ubiquitination, glycosylation, lipidation, oxidation, and disulfide bond formation (internal within a single protein or linking two protein/peptide chains together). Examples of common PTMs are provided in Figure \(\PageIndex{1}\ below.
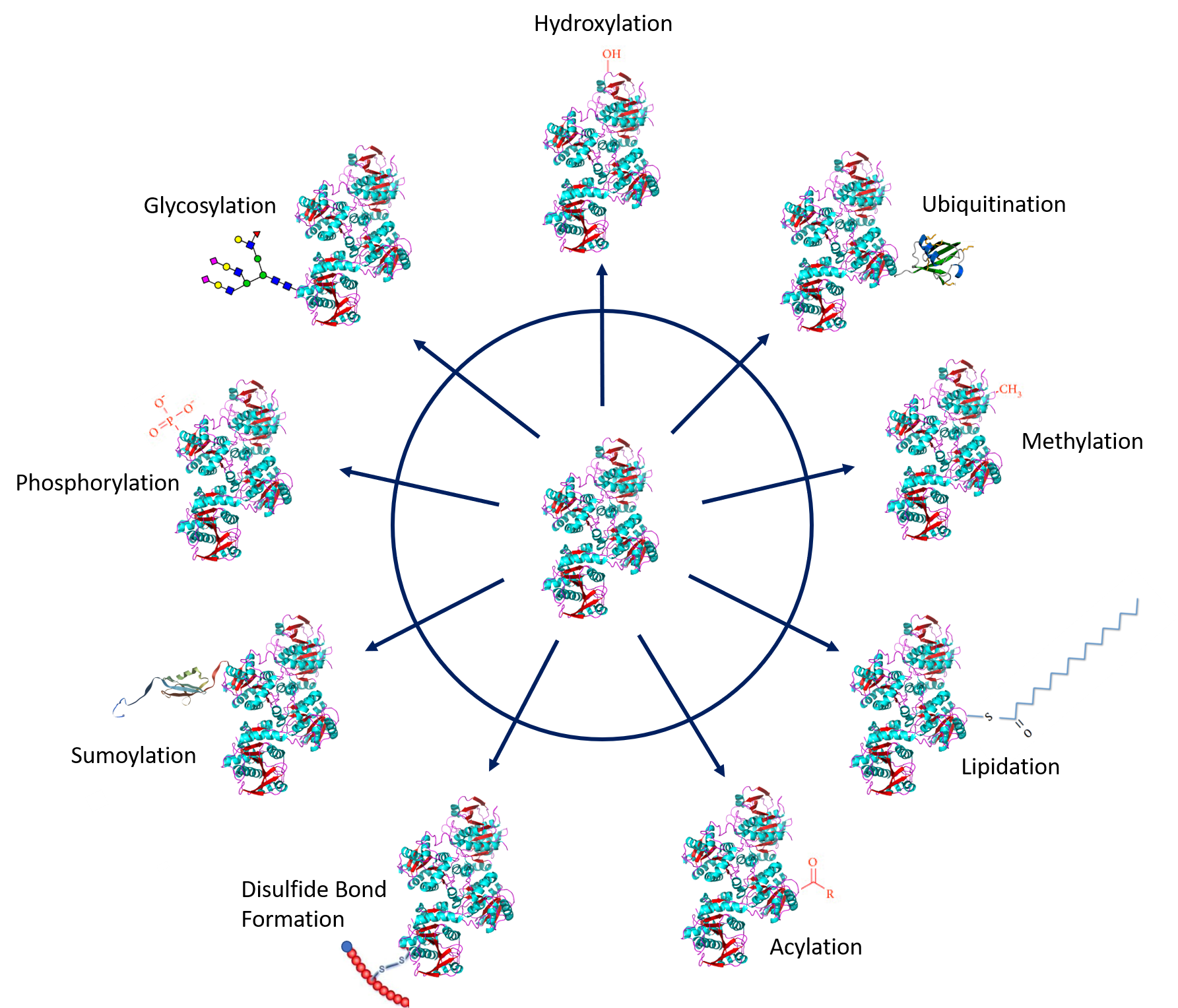
Protein Phosphorylation
One of the most common posttranslational modifications, protein phosphorylation, is the reversible addition of a phosphoryl group from adenosine triphosphate (ATP) to amino acid side chains such as serine, threonine, and tyrosine residues, as shown in Figure \(\PageIndex{2}\). This modification causes conformational changes that either (1) affect the catalytic activity to activate or inactivate the protein and/or cause the tendency of a protein to misfold and aggregate or (2) recruit other proteins to bind; both responses result in altered protein function and cell signaling. Phosphorylated proteins play critical, well-known roles in diverse cellular processes across eukaryotes, but phosphorylation also occurs in prokaryotic cells. In humans, about one-third of proteins are estimated to be substrates for phosphorylation. Indeed, phosphorylated proteins are now identified and characterized by high-throughput phosphoproteomics studies. The reversibility of protein phosphorylation is attributed to the actions of kinases and phosphatases, which phosphorylate and dephosphorylate substrates, respectively. The temporal and spatial balance of kinase and phosphatase concentrations within a cell mediates the size of its phosphoproteome.

Protein Acylation
The simplest form is N-acetylation of proteins. This occurs at the amino terminus amine and the ε-amino group of the lysine side chains through the action of acetylases. The acetylation of lysine side chains can be reversed through deacetylases (similar to the combined actions of protein kinases and phosphatases). Figure \(\PageIndex{3}\) shows the general reactions of protein N-acetylation. Interestingly, 80–90% of eukaryotic proteins are acetylated, yet the biological significance of this modification remains unclear.

The acetyl donor is acetyl-CoA. The N-terminus (reaction A) is acetylated by N-terminal acetyltransferases (NATs). The ε-amino group of lysine (reaction B) is acetylated by lysine acetyltransferases (KATs). The latter modification is reversed through two different enzymes. One is a simple hydrolysis (reaction C) catalyzed by lysine deacetylases (KDACs). The other (reaction D) requires NAD+ and is catalyzed by enzymes called sirtuins (silent information regulators). Since sirtuins use NAD+, one of the main oxidizing agents in the biological world, they link the protein acetylome with metabolic status and promote metabolic health.
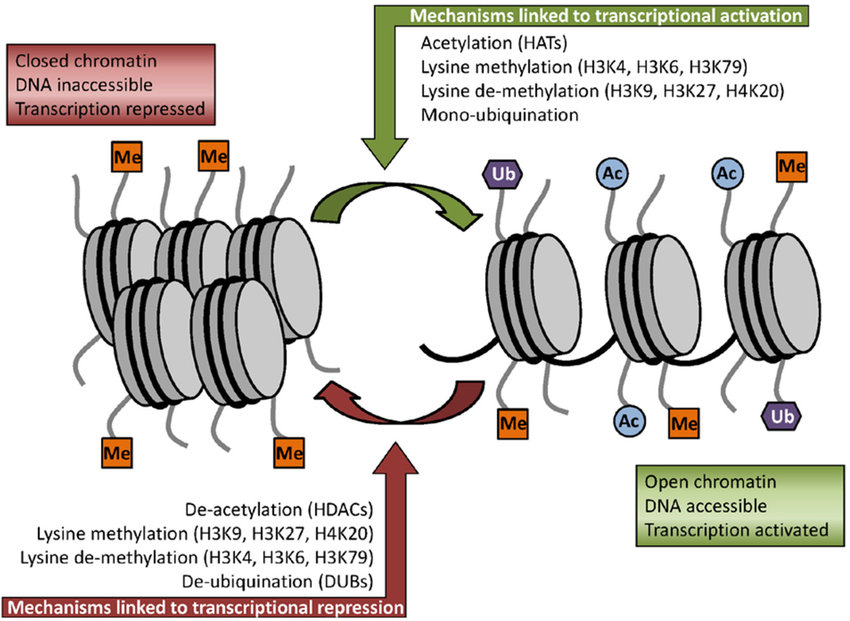
Histones, positively charged proteins found in eukaryotic cell nuclei, pack and order the DNA into structural units called nucleosomes, as shown in Figure \(\PageIndex{4}\).
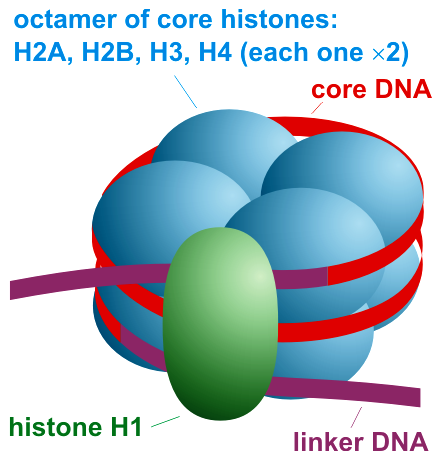
Histones contain multiple lysine and arginine residues that interact with the negatively charged phosphate groups of the DNA backbone. The nucleosome core comprises two H2A-H2B dimers and an H3-H4 tetramer, forming two nearly symmetrical halves by tertiary structure (C2 symmetry; one macromolecule is the mirror image of the other). The 4 'core' histones (H2A, H2B, H3, and H4) are structurally similar and highly conserved across evolution, all featuring a 'helix-turn-helix' motif (a DNA-binding protein motif that recognizes specific DNA sequences). They also share a feature of long 'tails' at one end of the amino acid structure, which is the site of post-translational modification, specifically N-acetylation.
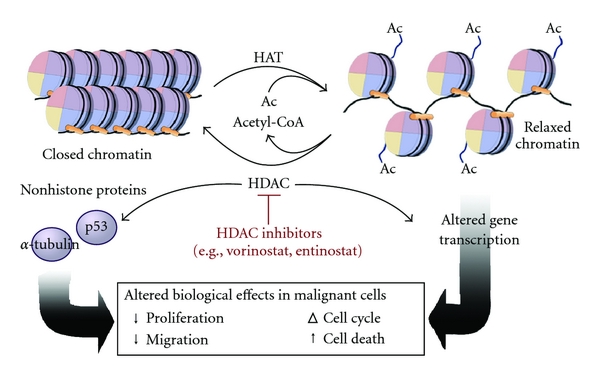
Histone acetylation typically results in transcriptional activation; deacetylation typically results in transcriptional suppression. Acetylation occurs via histone acetyltransferases (HATs) and is reversible via the action of histone deacetylases (HDACs). One group of histone deacetylases is the sirtuins (silent information regulator), which maintain gene silencing via hypoacetylation. Sirtuins have been reported to aid in maintaining genomic stability. Figure \(\PageIndex{5}\) shows the effect of acetylation on histone:DNA packing.
Although first described in histones, acetylation is also observed in cytoplasmic proteins. Acetylated proteins can also be modulated by cross-talk with other posttranslational modifications, including phosphorylation, ubiquitination, and methylation. Therefore, acetylation may contribute to cell biology beyond transcriptional regulation.
Protein Glycosylation
Protein glycosylation involves the addition of diverse sugar moieties to the protein core. Glycosylation significantly affects protein folding, conformation, distribution, stability, and activity. Glycosylated proteins can have additions of simple monosaccharides. For example, many nuclear transcription factors are modified in this way. Alternatively, some proteins are modified with highly branched polysaccharides, such as those on cell-surface protein receptors.
More than half of all mammalian proteins are believed to be glycosylated. While proteins exhibit improved stability and trafficking after glycosylation in vivo, glycan structures can alter protein functions or activities. Glycan structures are often modified by glycan-processing enzymes working within a cell at any given time. However, the structures are sometimes protein-specific, depending on protein trafficking properties and interactions with other cellular factors.
There are three types of protein glycosylation in higher eukaryotes: N-linked, O-linked, and C-linked. These types reflect their glycosidic linkages to amino acid side chains. In N-linked glycosylation, β-N-acetylglucosamine (GlcNAc) is attached through an amide linkage to the side chain of Asn within a consensus sequence of AsnXaaSer/Thr (Figure 8.8). N-linked glycans have multiple functions. While they act as ligands for glycan-binding proteins in cell-cell communication, they can also regulate plasma membrane glycoprotein aggregation and affect the half-lives of antibodies, cytokines, and hormones in serum.
O-linked glycosylation in higher eukaryotes occurs through several different mechanisms. The most abundant type of O-linked glycosylation is the mucin-type, which involves the attachment of an α-N-acetylgalactosamine (GalNAc) to the hydroxyl group of Ser/Thr side chains. Mucins are a family of high-molecular-weight, heavily glycosylated proteins (glycoconjugates) produced by epithelial tissues in most animals. A key characteristic of mucin proteins is their ability to form gels; therefore, they are a key component in most gel-like secretions, serving functions ranging from lubrication to cell signaling to the formation of chemical barriers. Aberrant expression of mucin-type O-linked glycans occurs in cancer cells and may provide targets for anticancer vaccines.
O-linked glycosylation, occurring with the addition of α-O-mannose, is the only form of O-linked glycosylation in yeast. Still, it also occurs in the brains of higher eukaryotes. Higher eukaryotes also have an α-O-fucose modification of Ser/Thr residues. This type of glycosylation modulates signaling pathways during eukaryotic development. Another modification, β-O-galactosylation, may contribute to the development of rheumatoid arthritis.
Finally, C-linked glycosylation involves the addition of α-mannose (Man) to the 2-position of the indole side chain of tryptophan residues. While first identified in ribonuclease 2, it also occurs on other proteins, including mucins, thrombospondin, and the Ebola virus soluble glycoprotein. Figure \(\PageIndex{6}\) shows examples of glycan adducts.

We will discuss glycoproteins in great detail in the next chapter.
Protein Ubiquitination and Sumoylation
Ubiquitination (or ubiquitylation) is the addition of an eight-kDa polypeptide, called ubiquitin, to lysine residues of target proteins via the C-terminal glycine residues of ubiquitin. Adding a single ubiquitin can lead to the addition of more ubiquitins to form ubiquitin chains on the target protein. The addition of one ubiquitin can modify the activity of the target protein. This can regulate a protein's activity, its location within the cell, and its interactions with other proteins. Adding a single ubiquitin to lysine 120 (K120) in humans and to K123 in yeast alters gene transcription by remodeling chromatin. However, if multiple ubiquitins are added to the target protein, forming a polyubiquitinated protein, the protein is tagged for degradation by the 26S proteasome. Figure \(\PageIndex{7}\) shows different patterns of ubiquitylation for proteins.

An elaborate system of enzymes attaches a ubiquitin through its C-terminal Gly 76 to an internal lysine (or cysteine) in a target protein. Then it adds more ubiquitins to form the structures shown above. Figure \(\PageIndex{8}\) shows the first step in the process, which requires three enzymes: E1, E2, and E3.

Once the first (or proximal) ubiquitin is added, more can be added to form chains. The steps involved in adding a second ubiquitin to the proximal one are shown in Figure \(\PageIndex{9}\).

Similarly, protein sumoylation is a reversible posttranslational modification in which a small ubiquitin-like modifier (SUMO) protein is covalently attached to a target protein. Like ubiquitin, SUMO is linked to a lysine side chain of target proteins and is removed by SUMO-specific isopeptidases. Sumoylation controls many aspects of nuclear function. Protein sumoylation is involved in many extranuclear neuronal processes and may be implicated in many neuropathological conditions.
Figure \(\PageIndex{10}\) shows ubiquitin (1UBQ, magenta) and SUMO-1 (1A5R, cyan) superimposed. Note the similarity in their structures even though they share only 18% sequence identity. Key amino acids are shown in spacefill.
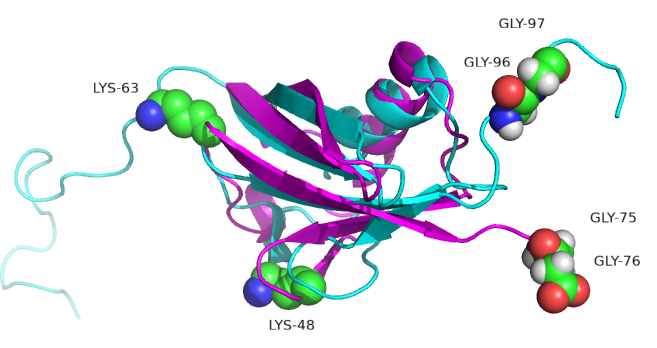
SUMO-1 doesn't target proteins for degradation. One of its roles is the transport of proteins between the nucleus and the cytoplasm, and it is involved in modulating protein-protein interactions. Both ubiquitin and SUMO-1 have a C-terminal glycine involved in isopeptide bond formation between ubiquitin or SUMO and the target protein. A significant difference between ubiquitin and SUMO-1 is along the disordered N-terminal end of SUMO. Lysines 48 and 63, found in ubiquitin, are also replaced by other amino acids. This may account for the observation that SUMO-1 does not form polymers.
The interplay between ubiquitylation and SUMOylation is complex. When protein ubiquitination is inhibited, SUMOylation of newly synthesized proteins increases. These can form phase-separated condensates called Promyelocytic Leukemia Nuclear Bodies (PML nuclear bodies), 0.1–1 μm condensates without a membrane found in most mammalian cell nuclei. These are somehow involved in nuclear gene expression.
Protein Oxidation
The reaction of proteins with a variety of free radicals and reactive oxygen species (ROS) leads to oxidative protein modifications such as the formation of protein hydroperoxides, hydroxylation of aromatic groups and aliphatic amino acid side chains, oxidation of sulfhydryl groups, oxidation of methionine residues, conversion of some amino acid residues into carbonyl groups, cleavage of the polypeptide chain, and formation of cross-linking bonds. Aromatic and sulfur-containing amino acid residues are particularly susceptible to oxidative modification.
Unless repaired or removed from cells, oxidized proteins are often toxic and can impair cellular viability, as they can form large aggregates. Oxidatively damaged proteins undergo selective proteolysis, primarily by the 26S proteasome, in a ubiquitin- and ATP-independent way. Upon extensive protein oxidation, these aggregates can become progressively resistant to proteolytic digestion, bind the 20S proteasome, and irreversibly inhibit its activity.
Protein carbonylation is defined as an irreversible posttranslational modification (PTM) whereby a reactive carbonyl moiety, such as an aldehyde, ketone, or lactam, is introduced into a protein. The first identified source of protein-bound carbonyls was metal-catalyzed oxidation (MCO). MCO results from the Fenton reaction when transition metal ions are reduced in the presence of hydrogen peroxide, generating highly reactive hydroxyl radicals. These hydroxyl radicals can oxidize amino acid side chains or cleave the protein backbone, leading to numerous modifications, including the formation of reactive carbonyls. For example, the oxidation of proline and arginine results in the production of glutamic semialdehyde, while lysine is oxidized to aminoadipic semialdehyde and threonine to 2-amino-3-ketobutyric acid. Direct oxidation of other amino acid residues can also lead to protein-bound carbonyls. Tryptophan oxidation by ROS produces at least seven oxidation products. Among them are kynurenine and N-formyl kynurenine and their hydroxylated analogs, which contain aldehyde or keto groups formed by oxidative cleavage of the indole ring.
Another important source of protein-bound carbonyls is reactive lipid peroxidation products formed during the oxidation of polyunsaturated fatty acids. Protein carbonylation can also occur via glycoxidation. Reactive α-carbonyls formed during glycoxidation, such as glyoxal, methylglyoxal, and 3-deoxyglucosone, can then modify the basic residues Lys and Arg to generate, for example, pyrralines and imidazolones. Glycation (i.e., the reaction of reducing sugars such as glucose or fructose with the side chains of lysine and arginine residues) forms Amadori and/or Hynes products. ROS can further convert these glycated residues into advanced glycation end products (AGEs), which carry carbonyl moieties.
Some oxidative modifications are made enzymatically and have key regulatory or structural functions within the modified proteins. For example, proline can be converted to hydroxyproline and lysine to hydroxylysine, as shown in Figure \(\PageIndex{11}\). 4-Hydroxyproline makes up about 13.5% of the residues within the mammalian collagen family of proteins. Recall that collagen is the main protein of connective tissue and represents about one-fourth of the total protein content in many animals. Hydroxyproline contributes to the stability of the triple helix and also aids in cross-linking collagen fibers to form larger macromolecular complexes.

Protein Methylation
Alkyl substituents are also a common posttranslational modification. Introducing such alkyl groups alters the hydrophobicity of the modified protein. The most common type of protein alkylation is protein methylation, which is mediated by methyltransferase enzymes. One-carbon methyl groups are added to nitrogen or oxygen (N- and O-methylation, resp.) on amino acid side chains, increasing protein hydrophobicity or neutralizing a negative charge when bound to carboxylic acids. While N-methylation is typically irreversible, O-methylation is potentially reversible. Methylation occurs so often that its primary methyl donor, S-adenosyl methionine (SAM), is one of the most-used enzymatic substrates after ATP. The methylation of a histone protein is shown in Figure \(\PageIndex{12}\).

This modification (not unlike phosphorylation reactions) plays a role in regulating protein-protein interactions. For instance, arginine methylation can either inhibit or promote protein-protein interactions, depending on the type of methylation. Protein methylation is also a common modification found in histone proteins. The transfer of methyl groups from S-adenosyl methionine to histones is catalyzed by enzymes known as histone methyltransferases. The N-terminal tails of histones H3 and H4 receive methyl groups on specific lysines. Methylation then determines if gene transcription is activated or repressed, thus leading to different biological outcomes.
Histone methylation was traditionally thought to be irreversible. However, histone demethylases demonstrate that this PTM is reversible. The simultaneous removal of one histone methylation and the addition of another can enable transcriptional fine-tuning.
In addition to methylation, histone acetylation, deacetylation, and mono-ubiquitination are essential parts of gene regulation, as shown below in Figure \(\PageIndex{13\). Acetylation removes the positive charge on the histones, thereby decreasing the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. Consequently, condensed chromatin is transformed into a more relaxed structure associated with greater gene transcription levels.
Nonhistone proteins also undergo methylation as a common PTM, which regulates signal transduction pathways. Furthermore, methylation works in concert with other PTMs to influence not only chromatin remodeling but also gene transcription, protein synthesis, and DNA repair.
Allosteric Regulation
Allosteric regulation fine-tunes most biological processes, including signal transduction, enzyme activity, metabolism, and transport. Allostery, an intrinsic property of a protein, is the regulation of activity at one site (also known as an orthosteric site) by a topographically and spatially distant site; the latter is designated as an allosteric site. Allosteric regulation occurs through binding a modulator (e.g., small molecule or protein) at an allosteric site to engender a conformational change that affects function at the orthosteric site. This effect may redistribute the conformational ensemble by either stabilizing an active conformation (allosteric activation) or destabilizing an inactive conformation (allosteric inhibition) in response to allosteric perturbations (Figure 8.13). Traditionally, the repertoire of allostery was primarily confined to determining the allosteric effects or mechanisms in individual multi-subunit or monomer proteins by conformational transitions. Recently, increasing evidence has indicated that allosteric signals can propagate across multiple proteins to sculpt allosteric networks.
Allosteric regulation is particularly important for the cell's ability to adjust enzyme activity in response to environmental conditions. Feedback control loops, such as feedback inhibition from downstream products or feedforward from upstream substrates, are common allosteric regulatory mechanisms found in nature. Another example of allostery involves oxygen binding to one of the subunits of hemoglobin, which prompts cooperative binding to other subunits.
We mentioned earlier that PTMs can induce conformational changes in proteins. Other methods to induce such allosteric changes include the binding of small molecules, the binding of proteins, changes in the redox environment of a protein that affect disulfide bonds, and general changes in protein flexibility and dynamics. Let's look at some examples of allostery brought about by these changes. Many of these examples are presented by Laskowski et al. ( https://doi.org/10.1016/j.febslet.2009.03.019).
Allosterism by small molecules: opening and closing active sites
Small-molecule binding at an allosteric site can induce small and large conformational changes in a distal active site. An often observed motion is a partial or full hinge-clamping conformational change that brings critical catalytic groups into a more organized, effective active site, thereby excluding water as a competing hydrolytic substrate (for example). One example of a small hinge-clamping change is seen in phosphoglycerate dehydrogenase (PGDH), which catalyzes the first step in serine synthesis (oxidation by NAD+). It converts 3-phosphoglycerate into 3-phosphohydropyruvate. The end product of the pathway, serine, binds to an allosteric site on PGDH and inhibits it, a classic example of feedback inhibition of the first reaction of a pathway by the end product. PGDH has a regulatory binding domain (RBD) that binds serine, a substrate-binding domain (SBD), an NAD+/NADH-binding domain (NBD), where the allosteric inhibitor binds, and binding domains for the substrate (SBD) and the NAD nucleotide (NBD). Figure \(\PageIndex{14}\) shows the small hinge-bending conformational change on bind conversion of monomeric apo-PGDH (1psd) to holo-PGDH with serine (not shown) bound in the RBD allosteric domain. The RBD-SBD domains rotate relative to the NBD domain by 15 °.
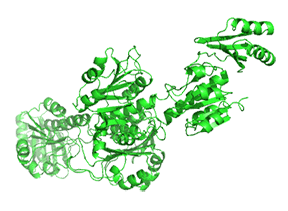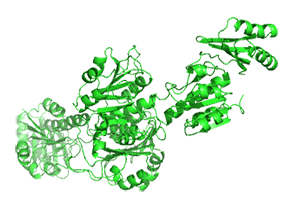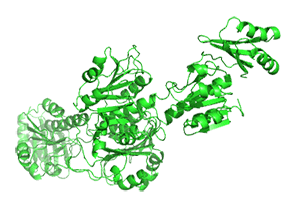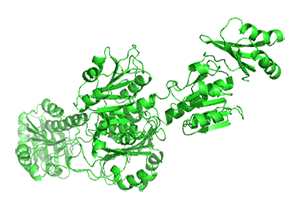
The functional enzyme is a tetramer (only the monomer is shown in the above figure). The rotation in the full tetramer aligns catalytic residues and closes off the active site.
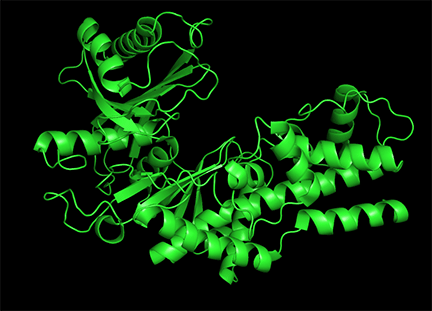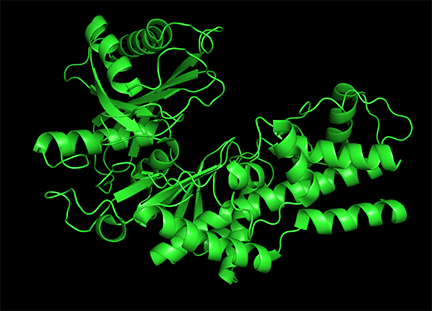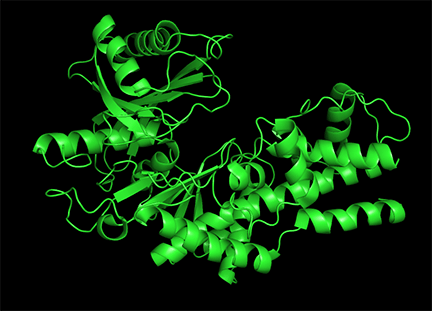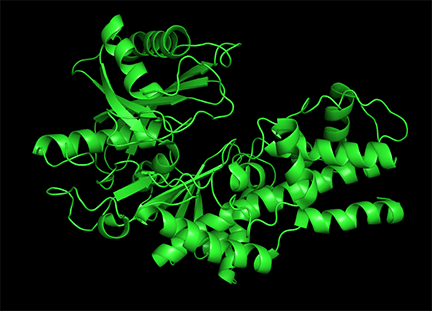
Conformational changes are ubiquitous in the binding of ligands to proteins. These changes also occur on substrate binding at the active site. It is important to recognize that substrate-induced changes in the active site are not allosteric since the conformational changes, no matter how big, are caused by binding at the active site, not at an allosteric site. A classic example of a large conformational change upon binding is the activation of hexokinase, the first enzyme in the glycolytic pathway, by glucose or glucose analogs. Figure \(\PageIndex{15}\) shows the large conformational change on binding a glucose analog to hexokinase (1hkg, no glucose; 2yhx, with glucose analog). The background is shown in black to remind readers that this is NOT an example of allosterism. The changes facilitate catalysis by excluding water, a competing substrate, from the active site after glucose and ATP bind (not shown). Upon binding the glucose analog in the active site, the site becomes sequestered ("jaw" clamping down on the binding) from solvent.
Allosterism by small molecules: Subtitle electrostatic effects
Often, no large conformation change is evident in the protein. In those cases, subtle rearrangements of key residues in the active site or near it (which might promote access) may result from allosteric effector binding. A simple change in the electrostatic environment might account for the effect. Such appears to be the case with chorismate mutase, an enzyme in the bacterial, fungal, and plant pathways for the synthesis of aromatic amino acids tyrosine and phenylalanine. The enzyme catalyzes the conversion of chorismate to prephenate, an intermediate in the synthesis of aromatic amino acids. An offshoot pathway takes chorismate to the last aromatic amino acid, tryptophan. The enzyme chorismate mutase is key in the metabolic decision pathway that leads to the production of tyrosine/phenylalanine or tryptophan. The enzyme is activated by tryptophan and inhibited by tyrosine. The aligned sequences of two crystal structures of the enzyme with the bound allosteric activator tryptophan (Trp 502 in pdb structure 1csm, showing a magenta glutamic acid 23) and with bound inhibitor (Tyr 300 in pdb structure 2csmA showing cyan Glu 23) are shown in Figure \(\PageIndex{16}\):
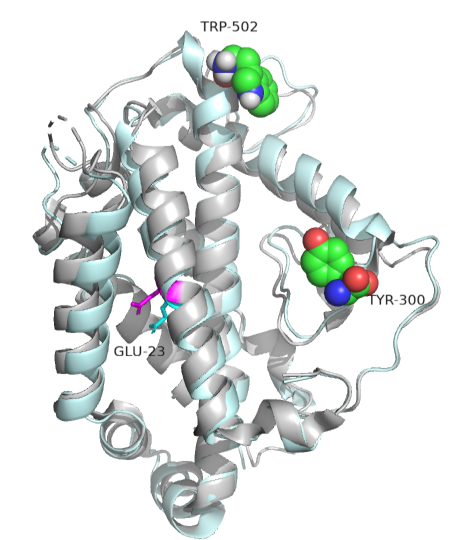
No significant changes occur in the protein's overall structure or active site. However, the alignment of glutamic acid 23 differs, which may underlie the observed allosteric effects. When active, glutamic acid 23 is buried in the active site pocket, but in the inhibited state, it swings into the binding site. Since chorismate is a charged dicarboxylic acid, Glu 23 probably repels it, inhibiting the enzyme's activity.
Allosterism by phosphorylation
We'll focus on one example of a large conformational change upon phosphorylation of a serine residue in glycogen phosphorylase (GP). This enzyme catalyzes the phosphorolysis of the acetal linkage between the terminal glucose of a glycogen polymer and the next glucose in the chain. This reaction is not a hydrolysis, in which water acts as a nucleophile to cleave acetal bonds. This is a key reaction in metabolism since it cleaves a major energy storage molecule, glycogen.
The enzyme is a multimer that exists in two major states, a T-state and an R-state glycogen phosphorylase (GP). We explored T and R states and allosterism in their interconversion when we discussed oxygen binding to hemoglobin in Chapter 5.3. Crystal structures of the T and R-state bound to allosteric activators IMP and AMP are known. High AMP concentrations imply low ATP concentrations and the need to mobilize glycogen reserves. Conversion to the R state is also promoted by the phosphorylation of Serine 14, which activates the enzyme. The allosteric activators AMP and IMP bind to a disordered loop (313-326) and change it to a form promoted by nucleotide binding. which is disordered in the free structure and adopts a conformation dictated mainly by the type of nucleotide that binds at this site. Figure \(\PageIndex{17}\) shows a monomer of the European rabbit glycogen phosphorylase in the R (active) state bound to AMP (3e3n).
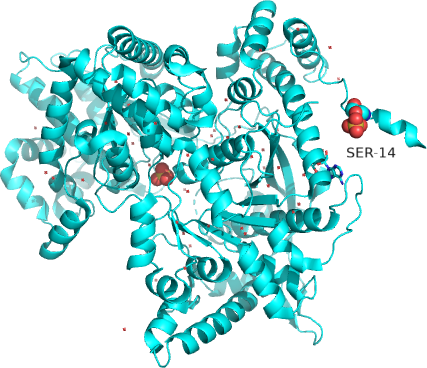
Only one chain of the active homodimer is shown. The helix containing the phospho-Ser 14 swings away from the rest of the protein.
Figure \(\PageIndex{18}\) shows the difference between the T (inactive) state of rabbit muscle glycogen phosphorylase (7P7D) and the R (active) state (3e3n, neither phospho-Ser 14 nor AMP shown). Note the huge conformational state elicited on phosphorylation of Ser 13.

Allosterism by small molecules: control of quaternary structure
The binding of small molecules at allosteric sites may promote or inhibit the formation of a protein's correct functional multimeric structure. One example is ATP phosphoribosyltransferase (APRT), which catalyzes the first step in synthesizing histidine. As we saw with phosphoglycerate dehydrogenase (PGDH), APRT has three domains. I and II comprise the active site that is located between them, and a regulatory domain, III (the functional protein is a dimer). As with PGDH, APRT is allosterically inhibited by the pathway's end product, histidine. On histidine binding to the inactive form (1nh7), a large conformational change results (1nh8), as shown in Figure \(\PageIndex{19}\).
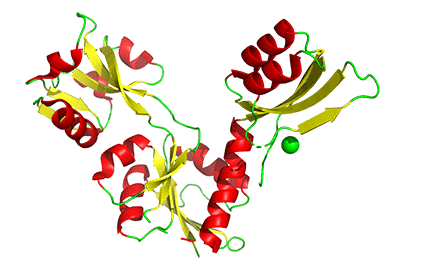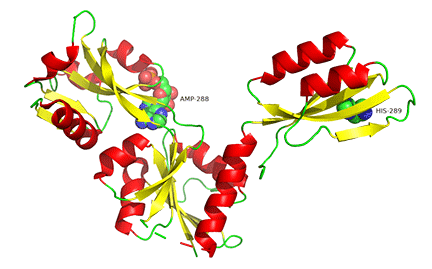
This change drives the formation of a hexamer - (dimer)3, which closes off the active site and inactivates the enzyme. The enzyme ribonuclease reductase behaves similarly.
Allosteric by protein binding
Another protein can bind to an enzyme and activate it by promoting an allosteric change. An example is the binding of a regulatory protein, CyclinA, to cyclin-dependent kinase 2 (CDK2). A large conformational change occurs upon cyclin binding to one side of CDK2's catalytic cleft, activating the enzyme by altering the active-site geometry and making it more accessible. CDK enzymes are involved in cell-cycle progression. For greater control of the cell cycle, cyclin binding and CDK2 phosphorylation are required for activity. Upon binding cyclin, the active site becomes more accessible for substrate (ATP) binding. Next, the amino acid that needs to be phosphorylated for activation, Thr 16, is made accessible. Figure \(\PageIndex{20}\) shows the conformational change in apo-CDK2 (1hc1) on cyclin binding, which allows ATP binding (1fin).

Cyclin (the regulatory protein) is shown in gray. ATP is shown in spacefill. CDK2 is shown as a colored cartoon.
Allosterism by disulfide bonds
It should make sense that cleaving an intrachain or interchain disulfide, whose presence constrains a specific protein conformation, should significantly alter protein structure and function. One example is the protein botulinum neurotoxin type A, a neurotoxin and Zn protease. It cleaves proteins such as synaptobrevin, syntaxin, and SNAP-25 in the neurosynapse, which are required for neurotransmitter release.
It has a catalytic (Zn peptidase/protease) and a translocation domain in a single protein chain, and contains two disulfide bonds when it is synthesized (PDB code 3bta). During cleavage at a specific peptide bond, the polypeptide is split into A and B chains, which remain connected by a single disulfide bond, with the A chain containing the catalytic domain. The B chain contains the translocation domain, which effectively still blocks the active site of the peptidase/protease domain. The disulfides are cleaved when the protein enters an endosome, which contains a more reducing environment. The separate A chain now separates from the inhibiting B domain and expresses protease activity. The active protein then enters the cytoplasm.
Figure \(\PageIndex{21}\) shows the domain structure and disulfide bonds of the unprocessed form. The N-terminal Zn proteinase domain (Peptidase_M27, amino acids 1-409) is followed by an intrachain disulfide between C429 and C 453.

The mature form is proteolyzed between amino acids 447 and 448 (within the sequences connected by the disulfide (429-453). The protein remains inactive until the disulfide bond is cleaved.
Figure \(\PageIndex{22}\) shows an interactive iCn3D model of botulinum neurotoxin type A (3bta).
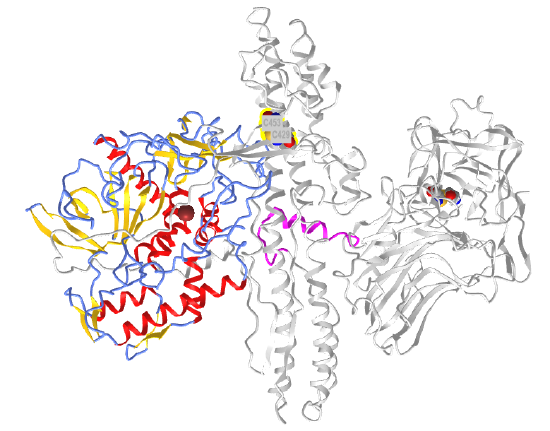
 Figure \(\PageIndex{22}\): Botulinum neurotoxin type A (3bta) (Copyright; author via source).
Figure \(\PageIndex{22}\): Botulinum neurotoxin type A (3bta) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...CfSsBH94jcPyb8
The N-terminal Zn peptidase/protease domain is shown in color, and the other regions of the protein are shown in gray. The two disulfide bonds are shown in spacefill, with CPK color. On cleavage of the disulfide bond between C429 and C453, the Zn protease domain, which is inhibited in the presence of the constraining disulfides, is released and activated.
Allosterism by changes in protein flexibility and population shifts
Sometimes, the crystal structure shows little difference between the apo- and holo-forms of a protein that contains a separate allosteric binding site. One example is the enzyme dihydrodipicolinate synthase (DHDPS), which catalyzes the first step in the bacterial and plant lysine biosynthetic pathways. DHDPS catalyzes the synthesis of 4-hydroxy-2,3,4,5-tetrahydro-2-dipicolinic acid from aspartate-β-semialdehyde and pyruvate. One explanation for allosterism in the protein is that it occurs on time scales too short to be seen in crystal or NMR structures. Rather, some transient conformation might cause allosterism. Such changes are detectable in the molecular dynamics simulation of the protein in the presence and absence of the allosteric effector molecule.
This possibility suggests that the allosteric effector might change the distribution of possible transient conformational states within a population of such states. If an allosteric effector bound one of the transient states in the population, that state would be "removed" from the ensemble of states, shifting the dynamic equilibrium among the populated states to produce more of the specific conformation that binds the allosteric effector. This sounds like the T to R state conversion in the MWC model oxygen binding to hemoglobin and the allosterism discussed above with the enzyme glycogen phosphorylase, only on a much shorter time scale. This model is consistent with a conformational selection model, in which the allosteric molecule binds preferentially to a preexisting, transient conformational state.
Zymogen Activation
A zymogen, also called a proenzyme, is an inactive precursor of an enzyme. A zymogen requires a biochemical change (such as a hydrolysis reaction that reveals the active site or a conformational change that exposes it) to become an active enzyme.
Protease enzymes secreted by the pancreas are initially synthesized as zymogens. The pancreas secretes zymogens to help prevent enzymes from inappropriately digesting proteins within the pancreatic cells where they are synthesized. Enzymes like Trypsin are synthesized as proenzymes. For trypsin, trypsinogen is an inactive precursor translated by the rough endoplasmic reticulum and transported to the Golgi apparatus for sorting. Trypsinogen is always co-synthesized and packaged with a pancreatic secretory trypsin inhibitor (PSTI), which inhibits premature activation. Thus, there are two mechanisms in place to maintain the inactivity of the protease within the pancreas: (1) synthesis of the zymogen or proenzyme form, and (2) co-expression of a trypsin inhibitor protein that will bind and inhibit any prematurely cleaved trypsin until it has reached the small intestine. An animation showing structural differences between bovine trypsinogen (magenta) with just amino acids 10-15 of the pre-sequence (spacefill orange) and mature trypsin (cyan) is shown in Figure \(\PageIndex{23}\).
During packaging in the Golgi apparatus, trypsinogen and other digestive enzymes condense into core particles and are packed into zymogen granules. The condensed enzymes are stable, and minimal activation occurs within the zymogen granules. Once pancreatic cells receive secretory stimuli, zymogen granules are released into the pancreatic duct lumen, which carries the digestive enzymes into the duodenum. Once in the duodenum, enteropeptidase activates trypsinogen by removing the 7-10 amino acids trypsinogen activation peptide (TAP) from the N-terminal region (see above). Removal of TAP induces a conformational change, resulting in active trypsin. TAP is immunologically distinct from the same sequence within trypsinogen, thereby allowing the detection of trypsinogen activation in situ.
Once activated, trypsin cleaves and activates other zymogen proteases and lipases in the duodenum. These include the activation of elastase, chymotrypsin, carboxypeptidase, and lipase, as shown in Figure \(\PageIndex{24}\). Zymogens are also found in other cellular processes. For example, caspases, intracellular proteases, are activated similarly during cellular apoptosis (programmed cell death). The process of blood clotting also involves the activation of zymogens.
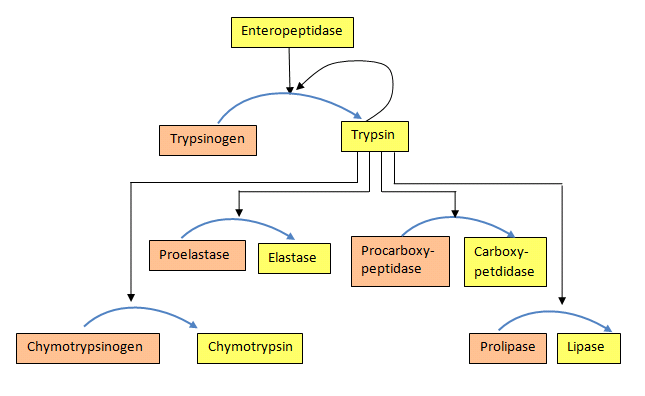
Figure \(\PageIndex{24}\): Activation of proenzymes
Isozymes
Isozymes (also known as isoenzymes) are enzymes that differ in amino acid sequence but catalyze the same chemical reaction. These enzymes usually display different kinetic parameters (e.g. different KM or Kcat values), or different regulatory properties. Isozymes enable fine-tuning of metabolism to meet the specific needs of a given tissue or developmental stage. Often, isozymes are encoded by homologous genes that are duplicated within the genome and then diverge over time. Isozymes should not be confused with allozymes, allelic variants of the same gene locus found within a population. Allozymes are enzymes encoded by different alleles of the same gene. Isozymes are enzymes from different genes that catalyze the same reaction, or enzymes from the same gene that produce, through differential splicing of primary RNA transcripts, sequences that are similar but different. We will focus on isozymes within this section. Part of the regulation of isozyme production can arise from differential expression.
Isozymes are usually the result of gene duplication. Over evolutionary time, if the function of the new variant remains identical to the original, then it is likely that one or the other will be lost as mutations accumulate, resulting in a pseudogene. However, if the mutations do not immediately prevent the enzyme from functioning, but instead modify either its function or its pattern of expression. The two variants may both be favored by natural selection and become specialized for different functions. For example, they may be expressed at different developmental stages or in different tissues. Some isozymes may also arise from convergent evolution and may not share a high degree of sequence homology or common ancestry.
The Cyclooxygenase I and II (Cox-1 and Cox-2) Isozymes
As an example of isozymes, we will discuss cyclooxygenases COX-1 and COX-2, which are also called Prostaglandin Synthases. They regulate a key step in prostaglandin and thromboxane synthesis and are the targets of nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin, ibuprofen, naproxen, and Celebrex (Figure \(\PageIndex{25}\)). Prostaglandins (PG) are a group of physiologically active lipid compounds called eicosanoids with diverse hormone-like effects in animals. Prostaglandins have been found in almost every tissue in humans and other animals. They are derived enzymatically from the fatty acid arachidonic acid. Every prostaglandin contains 20 carbon atoms, including a 5-carbon ring. They are a subclass of eicosanoids and the prostanoid class of fatty acid derivatives.

Cycloxygenases are a class of enzymes called dioxygenases, which incorporate both atoms of O2 into a substrate. We will explore the mechanism of cyclooxygenase in more detail in Chapter 13.5: Biological Oxidation and Reduction Reactions.
The structural differences between prostaglandins account for their different biological activities. A given prostaglandin may have different and even opposite effects in different tissues. The ability of the same prostaglandin to stimulate a reaction in one tissue and inhibit the same reaction in another tissue is determined by the type of receptor to which the prostaglandin binds. They act as autocrine or paracrine factors, with their target cells present near the site of their secretion. Prostaglandins differ from endocrine hormones in that they are produced at many sites throughout the human body rather than at a specific site and tend to act locally once secreted. Prostaglandins are implicated in various physiological processes, including gastrointestinal cytoprotection, hemostasis, thrombosis, and renal hemodynamics.
Through their role in vasodilation, prostaglandins are also involved in inflammation and can trigger the onset of a fever or the sensation of pain. They are synthesized in the walls of blood vessels. They prevent unnecessary clot formation and regulate smooth muscle contraction. The prostacyclins, a special class of prostaglandins, are powerful, locally acting vasodilators that inhibit platelet aggregation. Conversely, thromboxanes (produced by platelets) are vasoconstrictors and promote platelet aggregation. Their name comes from their role in the formation of clots (thrombosis).
The cyclooxygenases, COX-1 and COX-2, regulate the first two steps in prostaglandin synthesis and are bifunctional enzymes with two active sites. The first active site performs the bis-oxygenation and cyclization of arachidonic acid, whereas the second active site mediates a peroxidase (reduction) reaction to form PGH2. The enzyme is an example of a dioxygenase, which uses O2 as a substrate. The enzyme contains a heme cofactor. The functional enzymes of both COX-1 and COX-2 are homodimers of 70 kDa subunits, each containing an N-terminal epidermal growth factor domain, a membrane-binding domain, and a C-terminal catalytic domain. The cyclooxygenase active site is on the opposite side of the peroxidase active site in the catalytic domain.
Figure \(\PageIndex{26}\) shows an interactive iCn3D model of arachidonic acid bound to V349I murine COX-2 (6OFY). Just one chain of the dimer is shown.
.png?revision=1&size=bestfit&width=443&height=324)
Figure \(\PageIndex{26}\): Arachidonic acid and heme bound to V349I murine COX-2 (6OFY) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?gUtX3uAtwoozBvJUA
The heme is shown in stick, while the arachidonic acid is shown in spacefill. Note that arachidonic acid is very kinked, not extended, due to its four cis double bonds. Three additional amino acids that play key roles in NSAID inhibition of Cox are highlighted. These include Ser 530 (in model 531), which is covalently acetylated by aspirin, Arg 120 (in model 121), and Tyr 355 (in model 356). These play a key role in binding NSAIDs and in access to the arachidonic acid binding site.
The COX-1 enzyme is widely distributed in many tissues where it is constitutively expressed. Expression of the COX-2 isoform (shown in Figure \(\PageIndex{2}\)), on the other hand, is normally undetectable in most tissues (except for the central nervous system, kidneys, and seminal vesicles). Various inflammatory and mitogenic stimuli induce COX-2. A third isoform, COX-3, was recently identified as a COX-1 splicing variant. This new variant may play a role in processes such as fever and pain. Additionally, high COX-2 expression is observed in several forms of cancer. For example, COX-2 overexpression is related to poor prognosis in certain breast cancers and endometrial adenocarcinomas.
COX-2, unlike COX-1, is induced in inflammatory cells and activated by various inflammatory and mitogenic stimuli. Under these conditions, COX-2 activity leads to the production of prostanoid mediators that trigger important inflammatory processes. Although inflammation is initially a necessary process to fight infection, if it persists or remains uncontrolled, it can lead to chronic pathologies and tissue damage. This is why COX proteins are key targets for anti-inflammatory and pain management. Their actions are illustrated in Figure \(\PageIndex{27}\).

Figure \(\PageIndex{27}\): NSAID Inhibition of COX-1 and COX-2 Enzymes. The schematic representation of the COX-1 (large green figure) active site inhibited by a nonselective NSAID (central blue figure). The NSAID blocks the entrance channel to COX-1. The binding and transformation of arachidonic acid (bottom yellow figure) within COX-1 is prevented. Middle Lower Panel shows the inhibition of COX-2 by a nonselective NSAID (central blue figure). The nonselective binding uses an amino acid residue, Arg120, that is conserved in both enzymes. The right panel shows the inhibition of COX-2 by COX-2 selective NSAIDs (central red figure). The COX-2 side pocket allows specific binding of the COX-2 selective NSAIDs. The entrance channel to COX-2 is blocked. The bulkier COX-2-selective NSAID will not fit into the narrower COX-1 entrance channel, allowing COX-1 to remain active. Upper Figure by Saiz, M., Gonzalez, R., & Garcia, E., (2019) Protopedia and Lower Figure by Meek, I.L., et al. (2010) Pharmaceuticals 3(7):2146-2162.
Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Clinically available NSAIDs can be separated into three different classes based on their mechanism of action:
-
ASPIRIN: - Acts to irreversibly inhibit COX 1 & COX-2 by covalent acetylation of Ser 530 in the active site. Most notably, low doses of aspirin can suppress platelet COX-1 activity by 95% or more, an effect that is permanent for the lifetime of the platelet since platelets lack DNA and cannot synthesize new enzymes. Due to aspirin's antithrombotic properties at low doses, this treatment has been found to have cardioprotective effects and is often prescribed for patients at high risk of myocardial infarction. All other NSAIDs interact with COX isoforms reversibly and produce variable COX inhibition (ranging from 50% to 95%) in a time-dependent fashion based on how quickly they are metabolized in the body.
- NON-SELECTIVE COX INHIBITORS: Different non-selective NSAIDs have varying inhibitory effects against COX-1 & COX-2 (Figure 8.3). The two most commonly used over-the-counter drugs in this group (ibuprofen & naproxen) produce reversible platelet inhibition ranging from 50 to 95% in a reversible, time-dependent manner (see below). These NSAIDs may be insufficient to provide cardio-protection throughout a commonly used dosing interval and are not commonly used for this purpose. Ketorolac (Toradol ®), an NSAID most commonly used in a hospital setting to treat moderately severe pain, is classified as a non-selective NSAID. However, it is, arguably, a very selective COX-1 inhibitor (Figure 8.3). Inhibition of COX-1 can result in unwanted side effects, such as gastrointestinal discomfort and, in severe cases, ulceration.
-
COXIBS: Selective COX-2 inhibitors were designed and marketed to treat pain and inflammation while avoiding the GI side effects. However, soon after they were introduced into the market, their use led to the first reported incidence of increased cardiovascular events (myocardial infarction and stroke) in 2004. Rofecoxib (Vioxx ®), one of the most selective COX-2 inhibitors, was removed from the market because of mounting evidence for significant cardiovascular toxicity (Drazen, 2005). Celecoxib (Celebrex ®) is currently the only FDA-approved coxib available in the US, and it has been given a black box warning indicating the potential risk of cardiovascular toxicity. It has a 10-20-fold selectivity for COX-2 over COX-1. Etoricoxib (Arcoxia ®) is a second coxib with ~106-fold selectivity for COX-2 over COX-1 that is available outside the United States.

The structures of ibuprofen (Advil), naproxen, aspirin, Celebrex, and acetaminophen (Tylenol, which reduces fever and pain but not inflammation, so it's not an NSAID) are shown in Figure \(\PageIndex{29}\).

Although this extremely popular drug relieves pain and reduces fever, it does not reduce peripheral inflammation and is hence not classified as an NSAID. It does affect the central nervous system. It does not appear to bind the active sites of COX-1 or COX-2 (no PDB structures available), but it can reduce their activity, likely upstream or downstream of the pathways these enzymes participate in. It decreases prostaglandin production in the CNS, which reduces CNS pain and fever. It might act as a reducing substrate in the peroxidase site. Many proteins bind this drug. which is understandable given its relatively simple structure shown in Figure \(\PageIndex{5}\). A metabolite of it, p-aminophenol, can be esterified to arachidonic acid to produce the fatty acid amide, which can work through cannabinoid receptors to reduce pain.
Figure \(\PageIndex{30}\) shows an interactive iCn3D model of the NSAID ibuprofen bound to cyclooxygenase-2 (4PH9). Just one chain of the functional dimer is shown.
.png?revision=1&size=bestfit&width=442&height=293)
Figure \(\PageIndex{30}\): Ibuprofen bound to cyclooxygenase-2 (4PH9). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...syYYjSoeZRGNU8
Ibuprofen is sold as the racemic mixture of R and S enantiomers. Still, it was thought that only the S isomer's inhibition of Cox II led to its antiinflammatory effects, while the other isomer had no effects and no side effects. The crystal structures of IBP bound to both Cox I and II are known. At least in the mouse Cox2, the S-isomer binds more tightly than the R-isomer. Arg-120 and Tyr-355 at the entrance of the cyclooxygenase channel are important in the action of IBP in Cox2 (which are labeled in Figure \(\PageIndex{6}\).
As mentioned above, aspirin inhibits COXs through the covalent acetylation of a key serine side chain. The other NSAIDs inhibit prostaglandin H(2) synthase by blocking the channel for arachidonic acid binding. This should make you think they act as competitive inhibitors, which is true for ibuprofen and a methyl flurbiprofen derivative. However, the effect of two other NSAIDs, aclofenac and flurbiprofen, seems to have an added time component not found in the simple competitive inhibition model. They are called slow tight-binding inhibitors, and a simple model describing it is shown in Figure \(\PageIndex{31}\).

A time step suggests a conformational change after initial binding. Yet crystallographic studies of the four inhibitors mentioned above show that the NSAID-occupied active sites are essentially identical, suggesting no major global conformational change. It may be that the rate involves slow hydrogen bonding around two key residues, Arg 120 and Tyr 355.
All the enzyme inhibition models we discussed in Chapter 6.4 are based on reversible, rapid-equilibrium binding of an inhibitor to E (competitive), ES (uncompetitive), and E and ES (mixed/noncompetitive). Equations can be derived for slow-binding and slow-tight-binding inhibition. In these models, the degree of inhibition at a fixed inhibitor concentration changes with time to establish an "equilibrium" among all species. The model above works for both. Both types of inhibitors (slow and slow-tight) inhibit enzymes in a time-dependent fashion. In slow binding, it takes longer to establish equilibrium among the three species, E, EI, and EI*. In slow binding, the steps determining the concentration of EI (k3I and k-3) are assumed to be fast compared to those involved in determining the concentration of EI* (k4 and k-4). When [I] >> [E], the following dissociation constants for pure competitive (KIS = KC) and for the slow binding inhibition (KI), which accounts for mass balance, hold.
\begin{equation}
\mathrm{K}_{\mathrm{IS}}=\mathrm{K}_{\mathrm{C}}=\frac{[\mathrm{E}][\mathrm{I}]}{[\mathrm{EI}]}
\end{equation}
\begin{equation}
\mathrm{K}_{\mathrm{I}}^{*}=\frac{[\mathrm{E}][\mathrm{I}]}{[\mathrm{EI}]+\left[\mathrm{EI}^{*}\right]}
\end{equation}
In initial rate Michaelis-Menten kinetics, v0 is measured as a function of [S]. The initial rate, v0, is determined by measuring [P] vs. t at the beginning of the reaction, when the substrate is not depleted. Valid v0 values require that [P] vs t curves are linear. The slope of that line is the initial velocity, v0. This is true for uninhibited and reversibly inhibited reactions (competitive, uncompetitive, and mixed). However, with slow binding inhibition, the [P] vs t curves bend with time as the slower formation of EI* occurs. It's a bit like the pre-steady state assumption in enzyme kinetics. At very short times (msec), the P vs. t curves bend as a steady state emerges. It's the same with slow-binding inhibition. This is illustrated in Figure \(\PageIndex{32}\).

A progress curve equation showing [P] vs. t for slow binding in inhibition is shown below without inhibition.
\begin{equation}
[\mathrm{P}]=\mathrm{v}_{\mathrm{s}} \mathrm{t}+\frac{\left(\mathrm{v}_{\mathrm{o}}-\mathrm{v}_{\mathrm{s}}\right)\left(1-\mathrm{e}^{-\mathrm{k}_{\mathrm{a}} \mathrm{t}}\right)}{\mathrm{k}_{\mathrm{a}}}+\mathrm{C}
\end{equation}
In this equation, ka is the apparent first-order rate constant for the development of the steady state at a given substrate and inhibitor concentration, v0 is the very first initial rate, and vs is the steady-state rate.
Slow tight-binding inhibition occurs when the rate constants for net production of EI* are fast compared to the step for forming EI. In this case, even small amounts of I will produce EI* as the reaction is pulled to EI*, and more complicated equations must be used to determine product vs time equations.
Other chemical models could also account for slow-binding inhibition. These include a slow binding rate constant (k4), a slow isomerization of EI after fast binding of I, or a slow binding to a specific, low-population conformation of the enzyme, in a process called conformational selection. Slow-binding enzyme inhibitors (such as acetylcholinesterase) are known.
Here is a Vcell simulation showing progress curves for an uninhibited reaction, one in the presence of a competitive inhibitor, and one in the presence of a slow (competitive) inhibitor.
 MODEL
MODEL
Comparison No Inhibition, Competitive Inhibition, and Slow (Competitive) Inhibition
Vcell reaction diagram with all equations based on mass action (not Michael-Menten kinetics)

Concentrations
- A, B and C (substrates), t0 = 1 uM
- I1 and I2 (inhibitors) = 1 uM
- E, E1 and E2 (enzymes) at t0 = 0.1 uM
- P, Q and R (products) at t0 = 0 uM
Rate Constants
All forward (kf) and reverse (kr) rate constants are set initially to 1 except for
- kr2 for all reactions = 0 and kf4 = 1.
Select Load [model name] below
Select Start to begin the simulation.
Only plots of P, Q and R (products) are initially shown. Select Plot to change Y axis min/max, then Reset and Play | Select Slider to change which constants are displayed | Select About for software information.
Move the sliders to change the constants and see changes in the displayed graph in real-time.
Time course model made using Virtual Cell (Vcell), The Center for Cell Analysis & Modeling, at UConn Health. Funded by NIH/NIGMS (R24 GM137787); Web simulation software (miniSidewinder) from Bartholomew Jardine and Herbert M. Sauro, University of Washington. Funded by NIH/NIGMS (RO1-GM123032-04)
Coxibs and the Thromboxane/Prostacyclin Imbalance Hypothesis
Previous research indicates that in the cardiovascular system, greater inhibition of COX-2 vs. COX-1 (as produced by COX-2-selective “coxibs”) can tip the normal balance between prostacyclin & thromboxane, increasing the likelihood of platelet aggregation and vasoconstriction. These effects can help to explain the higher incidence of myocardial infarction and stroke observed when these drugs have been used clinically. The mechanisms involved are illustrated in Figure \(\PageIndex{33}\).

The left graphic illustrates the normal balanced effect between prostacyclin (PGI2) and Thromboxane (TXA2). PGI2 is produced primarily by COX-2 activity in the endothelial cell wall of blood vessels. PGI2 produces vasodilation and inhibits platelet activation. In contrast, TXA2 is produced primarily by COX-1 activity in platelets, leading to vasoconstriction and enhanced platelet aggregation. Normal vascular homeostasis is maintained when the effects of PGI2 & TXA2 are balanced. However, when the balance is tipped toward TXA2 formation after selective COX-2 inhibition (right graphic), vasoconstriction and platelet clumping are more likely to occur, potentially increasing the risk of cardiovascular events such as myocardial infarction and stroke. Overall, the COX-1/COX-2 isozyme example sheds light on the complexity of biological systems and the ability of slight adjustments in gene expression to generate varied, tissue-specific responses.
Summary
(Summary written by Claude, Sonnet 4.6, Anthropic)
This chapter examines the major mechanisms by which protein activity is regulated after synthesis, spanning covalent post-translational modifications, allosteric conformational changes, zymogen activation, and isozyme-based tissue-specific control — illustrating that protein function in cells is regulated by multiple overlapping mechanisms simultaneously and that even subtle structural differences between isozymes can have profound pharmacological consequences.
Post-translational modifications (PTMs) dramatically amplify proteome complexity: approximately 20,000 human protein-coding genes produce ~145,000 transcripts through alternative splicing (~7.4 transcripts per gene), and PTMs then generate potentially 1 million distinct proteoforms. PTMs involve the enzymatic or nonenzymatic transfer of a chemical group (typically from an electrophilic co-substrate to a nucleophilic amino acid side chain) and include phosphorylation, acetylation, methylation, glycosylation, ubiquitination, sumoylation, and oxidative modifications. Phosphorylation of Ser, Thr, and Tyr by kinases (reversed by phosphatases) is among the most common post-translational modifications, affecting ~1/3 of human proteins; it alters protein conformation, activity, protein-protein interactions, and aggregation propensity. Acetylation at the α-amino terminus and at Lys ε-amino groups (transferred from acetyl-CoA by NATs and KATs; reversed by KDACs and NAD⁺-dependent sirtuins, connecting acetylation state to metabolic status) affects ~80–90% of eukaryotic proteins. Methylation by methyltransferases using SAM as the methyl donor is the second-most-used enzymatic substrate, after ATP. Glycosylation (N-linked to Asn, O-linked to Ser/Thr, C-linked to Trp) affects over half of mammalian proteins and influences folding, stability, trafficking, and cell-cell recognition. The ubiquitin-proteasome system uses a cascade of E1, E2, and E3 enzymes to append ubiquitin via its C-terminal Gly 76 to Lys residues on target proteins; polyubiquitin chains (particularly K48-linked) target proteins for degradation by the 26S proteasome, while monoubiquitination regulates activity, localization, and interaction with other proteins without triggering proteolysis. SUMO (small ubiquitin-like modifier) shares the β-grasp fold with ubiquitin despite only ~18% sequence identity, attaches by the same isopeptide linkage to Lys residues via analogous E1/E2/E3 enzymes, but regulates nuclear transport and protein-protein interactions rather than directing proteasomal degradation. Among the most biologically impactful PTMs are histone modifications in the nucleus: Lys acetylation by HATs neutralizes the positive charge on histone tails, weakening electrostatic interaction with negatively charged DNA phosphates, relaxing chromatin structure, and promoting transcription (reversed by HDACs and sirtuins); Lys and Arg methylation by histone methyltransferases either activates or represses transcription depending on the site and degree of methylation; and monoubiquitination of specific histone Lys residues contributes to chromatin remodeling.
Allosteric regulation fine-tunes enzyme activity in response to diverse signals without permanently altering protein structure. The mechanistic diversity is striking. In phosphoglycerate dehydrogenase, the pathway end product, serine, binds to a regulatory domain and induces a 15° hinge-clamping rotation of the active-site domain relative to the NAD⁺-binding domain, closing the active site—a classic example of feedback inhibition that shows how small-molecule allosteric effectors can produce large functional effects through modest geometric changes. In chorismate mutase, allosteric activation by tryptophan and inhibition by tyrosine occur without detectable global conformational change; instead, the key electrostatic residue Glu 23 repositions between buried (active state) and swung-into-the-active-site (inhibited state) orientations, electrostatically repelling the charged dicarboxylate substrate. In glycogen phosphorylase, phosphorylation of Ser 14 by phosphorylase kinase drives a massive T→R conformational transition that swings a helix out from the protein core, reorganizes the active site, and activates the enzyme; high [AMP] (signaling low energy) achieves the same T→R shift by binding to the allosteric nucleotide site. In ATP phosphoribosyltransferase, histidine binding to the inactive dimer drives a large conformational change that promotes hexamerization, closing all active sites simultaneously and providing ultrasensitive feedback inhibition of the first step of histidine biosynthesis. In botulinum neurotoxin, reduction of a disulfide bond that connects an inhibitory translocation domain to the catalytic Zn protease domain is the switch that releases the active protease in the reducing environment of the endosome. And in some enzymes (e.g., DHDPS), allosteric effects are detectable only through molecular dynamics simulations, which reveal changes in the population of transient conformational states upon effector binding — a conformational selection mechanism operating on microsecond timescales, faster than crystallographic resolution.
Zymogen activation provides irreversible, triggered regulation appropriate for contexts requiring sharp on/off control. Pancreatic digestive proteases are synthesized in the acinar cells as inactive zymogens — trypsinogen, chymotrypsinogen, proelastase, procarboxypeptidase — and co-packaged with the pancreatic secretory trypsin inhibitor (PSTI) to prevent inadvertent autodigestion. Once released into the duodenum, enteropeptidase cleaves trypsinogen's N-terminal 7–10 residue activation peptide (TAP), triggering a conformational change that reveals the active site and generates active trypsin. Trypsin then autocatalytically activates more trypsinogen and proteolytically activates all other pancreatic zymogens in a cascade — a cascade architecture that provides both amplification and spatial control. The same principle applies to blood coagulation (a cascade of clotting proteases) and apoptosis (activation of procaspases).
COX-1 and COX-2 isozymes provide a medically important illustration of how isozymes arising from gene duplication and divergence can acquire tissue-specific expression patterns and distinct regulatory properties while retaining the same catalytic reaction. Both are bifunctional homodimers (70 kDa subunits) with cyclooxygenase and peroxidase activities, heme cofactors, and the same fundamental mechanism (bis-oxygenation of arachidonic acid at the cyclooxygenase site, reduction at the peroxidase site, producing PGH₂, the precursor to prostaglandins, prostacyclins, and thromboxanes). COX-1 is constitutively expressed and produces PGI₂ and TXA₂ for homeostatic functions (gastric cytoprotection, renal hemodynamics, hemostasis). COX-2 is normally undetectable in most tissues but is strongly induced by inflammatory cytokines and growth factors; its overexpression is found in cancers, including breast and endometrial adenocarcinoma. A single amino acid difference (Val 523 in COX-1 vs. Ile 523 in COX-2) creates a side pocket in the COX-2 active site channel that can accommodate the bulkier sulfonamide/sulfone groups of coxibs but is too narrow for them in COX-1, providing the structural basis for COX-2 selectivity. NSAIDs act by three mechanisms: aspirin irreversibly acetylates Ser 530, permanently blocking the arachidonic acid channel (in platelets, which lack nuclei, this effect is permanent for the platelet's lifetime, forming the basis of low-dose aspirin cardioprotection); non-selective NSAIDs (ibuprofen, naproxen, ketorolac) reversibly block the channel through interactions with Arg 120 and Tyr 355; and coxibs exploit the COX-2 side pocket for selective inhibition. The cardiovascular toxicity of COXibs — the basis for rofecoxib's withdrawal and celecoxib's black box warning — is explained by the thromboxane/prostacyclin imbalance hypothesis: selective COX-2 inhibition reduces PGI₂ production in vascular endothelium (vasodilatory, antiplatelet) without reducing platelet TXA₂ (vasoconstrictive, proplatelet), tipping the balance toward thrombosis and explaining increased myocardial infarction and stroke risk.
Some References
- Shen, Q., Wang, G., Li, S., Liu, X., Lu, S., Chen, Z., Song, K., Yan, J., Geng, L, Huang, Z., Huang, W., Chen, G., and Zhang, J. (2016) ASDv3.0: Unraveling allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Research 44(D1):D527-D535. Available at: https://academic.oup.com/nar/article/44/D1/D527/2503129
- Wikipedia contributors. (2020, January 19). Isozyme. In Wikipedia, The Free Encyclopedia. Retrieved 03:42, May 19, 2020, from https://en.Wikipedia.org/w/index.php?title=Isozyme&oldid=936548836
- Wikipedia contributors. (2020, April 30). COX-2 inhibitor. In Wikipedia, The Free Encyclopedia. Retrieved 07:00, May 22, 2020, from en.Wikipedia.org/w/index.php?title=COX-2_inhibitor&oldid=954080651
- Clarkson, C.W. (2018) Major Side Effects of NSAIDs and COX-2 Selective Inhibitors. TUSOM Pharmwiki. Available at: http://tmedweb.tulane.edu/pharmwiki/doku.php/nsaid_side_effects?do=
- Santos, A.L. and Lindner, A.B. (2017) Protein Porsttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Medicine and Cellular Longevity, Article ID: 5716409. Available at: https://www.hindawi.com/journals/omcl/2017/5716409/#copyright
- Wikipedia contributors. (2020, May 12). Protein phosphorylation. In Wikipedia, The Free Encyclopedia. Retrieved 17:38, May 25, 2020, from en.Wikipedia.org/w/index.php?title=Protein_phosphorylation&oldid=956228409
- Szylveszter, K.P., Németh, T., and Mócsai, A. (2019) Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 10.3389(2019.01862). Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01862/full
- Wikipedia contributors. (2020, May 7). Histone. In Wikipedia, The Free Encyclopedia. Retrieved 21:12, May 25, 2020, from en.Wikipedia.org/w/index.php?title=Histone&oldid=955458038
- Wikipedia contributors. (2020, May 13). Mucin. In Wikipedia, The Free Encyclopedia. Retrieved 01:10, June 7, 2020, from en.Wikipedia.org/w/index.php?title=Mucin&oldid=956387296
- Wikipedia contributors. (2020, March 15). Allosteric regulation. In Wikipedia, The Free Encyclopedia. Retrieved 04:07, June 7, 2020, from en.Wikipedia.org/w/index.php?title=Allosteric_regulation&oldid=945637073
- Dixit, Ajay. Dawra, Rajinder K. Dudeja, Vikas. Saluja, Ashok K. (2016). Role of trypsinogen activation in genesis of pancreatitis.
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.25 - Coll-Martinez, B., and Crosas, B. (2019) How the 26S Proteasome Degrades Ubiquitinated Proteins in teh Cell. Biomolecules 9(9):395. Retrieved from: https://www.mdpi.com/2218-273X/9/9/395
- Liu, W., Tang, X., Qi, X., Ghimire, S., Ma, R., Li, S., Zhang, N., and Si H. (2020) The Ubiquitin Conjugating Enzyme: An Important Ubiquitin Transfer Platform in Ubiquitin-Proteosome System. Int J Mol Sci 21(8):2894. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7215765/