
12.4: Biological Oxidation-Reduction Reactions
- Page ID
- 15000


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Understand the Fundamentals of Oxidation Reactions:
- Describe the general steps of oxidation processes in organic chemistry, including initiation, propagation (and branching), and termination reactions.
- Compare the mechanisms of combustion (using dioxygen) with those of controlled oxidation using alternative oxidizing agents like permanganate and chromate.
-
Contrast Chemical and Biological Oxidizing Agents:
- Explain why dioxygen is often unsuitable for controlled oxidation in organic synthesis and how alternative oxidizing agents overcome these challenges.
- Identify the unique challenges of oxidizing organic molecules in biological systems and the specialized oxidizing agents (e.g., NAD⁺, FAD, monooxygenases) that are employed instead.
-
Examine the Chemistry and Function of NAD⁺ and FAD:
- Illustrate the structures of NAD⁺ (and its reduced form, NADH) and FAD (and FADH₂), and explain how their chemical properties make them effective two-electron hydride carriers.
- Discuss the mechanistic basis of hydride transfer reactions catalyzed by dehydrogenases, and how these reactions integrate into metabolic pathways (e.g., conversion of pyruvate to lactate).
-
Explore the Roles of Dehydrogenases in Biological Oxidation:
- Describe how dehydrogenase enzymes use NAD⁺ and FAD to oxidize substrates and drive energy production in pathways such as glycolysis and the Krebs cycle.
- Use standard reduction potentials and the Nernst equation (ΔG = –nFE°) to analyze the thermodynamics of these redox reactions.
-
Differentiate Among Enzyme Classes that Utilize Dioxygen:
- Compare monooxygenases (including hydroxylases) that incorporate one oxygen atom into substrates, dioxygenases that incorporate both atoms, and oxidases that transfer electrons to dioxygen without substrate incorporation.
- Illustrate the roles of these enzyme classes in diverse biological contexts, such as xenobiotic metabolism, hormone synthesis, and detoxification.
-
Delve into the Mechanism and Function of Cytochrome P450 Enzymes:
- Outline the catalytic cycle of cytochrome P450s, emphasizing the role of the heme cofactor, electron transfer from NADPH via cytochrome P450 reductase, and the activation of dioxygen for substrate hydroxylation.
- Analyze how cytochrome P450-mediated reactions can both detoxify substrates and, paradoxically, activate procarcinogens.
-
Understand the Structural Basis for Redox Tuning in Enzymes:
- Explain how the protein microenvironment (e.g., ligand identity, hydrophobicity, hydrogen bonding) modulates the redox potential of enzyme-bound cofactors such as FAD and metal centers in cupredoxins like azurin.
- Evaluate examples where mutations alter redox potential and discuss the implications for enzyme function.
-
Recognize the Sequential Oxidation of Organic Molecules in Metabolism:
- Describe the stepwise oxidation of methane to carbon dioxide in methanotrophs, including the enzymes involved in each oxidation step (e.g., methane monooxygenase, methanol dehydrogenase).
- Discuss how these sequential reactions illustrate the controlled use of oxidizing agents to extract energy from organic molecules.
-
Integrate the Role of Heme Proteins in Diverse Redox Processes:
- Compare the functions of different heme proteins (hemoglobin, cytochrome C, cytochrome P450) and explain how variations in coordination and protein environment enable them to carry out distinct redox roles.
- Link structure to function by discussing how differences in heme ligation (e.g., proximal histidine vs. cysteine) determine each protein’s reactivity and physiological role.
-
Explore Transition Metal Homeostasis and Its Impact on Oxidative Reactions:
- Discuss the importance of metalloproteins in binding, transporting, and regulating transition metals such as Fe, Zn, and Cu.
- Explain how differences in metal binding affinities and coordination geometries are critical for preventing metal-induced oxidative damage while facilitating essential redox reactions.
These learning goals are intended to help students integrate chemical principles with biological context, enabling a deeper understanding of how oxidizing agents function in both laboratory settings and living systems.
General Oxidizing Agents
Before considering common biological oxidizing agents, look back at the ones you saw in other chemistry classes. Oxidizing agents are required to oxidize organic molecules. In chemistry labs, you likely never used dioxygen as an oxidizing agent. It is difficult to limit the extent of oxidation using dioxygen. In addition, side reactions are likely given the nature of the reactive oxygen reduction products. The mechanisms of combustion reactions of organic molecules with dioxygen to produce carbon dioxide and water are complicated. Table \(\PageIndex{1}\) shows some key steps in the combustion of methane.
Initiation
\[\ce{CH4 → CH3^{.} + H^{.}} \nonumber\]
\[\ce{O2 → 2 O^{.}} \nonumber\]
Propagation
\[\ce{CH4 + H^{.} → CH3^{.} + H2} \nonumber\]
\[\ce{CH4 + HO^{.} → CH3^{.} + H2O} \nonumber\]
\[\ce{CH3^{.} + O^{.} → CH2O + H^{.}} \nonumber\]
\[\ce{CH2O + HO^{.} → CHO^{.} + H2O} \nonumber\]
\[\ce{CH2O + H^{.} → CHO^{.} + H2} \nonumber\]
\[\ce{CHO^{.} → CO + H^{.}} \nonumber\]
\[\ce{CO + HO^{.} → CO2 + H^{.}} \nonumber\]
Branching
\[\ce{H^{.} + O2 → HO^{.} + O^{.}} \nonumber\]
Termination
\[\ce{H. + R^{.} + M → RH + M^{*}} \nonumber\]
In chemistry, other oxidizing agents are often used, including permanganate and chromate. Their mechanism of action is illustrated in Figure \(\PageIndex{1}\).


Oxygen can often be inserted into a molecule in a nonoxidative process by hydration of an alkene to an alcohol (a readily reversible reaction), which could then be oxidized to either an aldehyde/ketone or carboxylic acid using an appropriate oxidizing agent.
Most biological oxidation reactions (such as those found in glycolysis, the Krebs cycle, and fatty acid oxidation) do not use dioxygen as the immediate oxidizing agent. Instead, they use nicotinamide adenine dinucleotide (NAD+) or flavin adenine dinucleotide (FAD) as oxidizing agents, which are reduced. Enzymes that use these oxidizing agents are usually called dehydrogenases. Dioxygen can also introduce oxygen atoms into biological molecules in oxidative reactions. Enzymes that introduce one oxygen atom of dioxygen into a molecule (and the other oxygen into water) are called monooxygenases. (Note: some monooxygenases that hydroxylate biomolecules are called hydroxylases.) Those that introduce both atoms of dioxygen into a substrate are called dioxygenases. These oxygenases are not usually used to oxidize organic molecules for energy production. Instead, they introduce O atoms for other reasons, including increasing the solubility of nonpolar aromatics to facilitate secretion and produce new molecular species with different biological activities. Finally, dioxygen can oxidize biological molecules without adding oxygen atoms to the substrate. Instead, electrons lost from the oxidized substrate are passed via intermediate electron carriers to dioxygen, which gets reduced to superoxide (if one electron is added), hydrogen peroxide (if two electrons are added), or water (if 4 electrons are added). These enzymes are called oxidases. (Note: The letters oxi- or oxygen- are used in all the enzymes that use dioxygen as the oxidizing agent.)
In this chapter section, we will discuss biological oxidation reactions. Most introductory biochemistry texts don't approach oxidation reactions in one cohesive chapter. Probably because of that, when I was learning biochemistry, I found the presentation of these different enzymes involved in redox reactions very confusing. Hopefully, this section will alleviate that problem. First, the chemistry of NAD+ and FAD will be discussed. Then, the enzymes using dioxygen in oxidative reactions (monooxygenases, dioxygenases, and oxidases) will be explored.
The Chemistry of NAD+ and FAD
NAD+ is a nicotinic acid or nicotinamide derivative, as illustrated in Figure \(\PageIndex{2}\).

Figure \(\PageIndex{2}\): Structure of niacin derivatives
It and its reduction product, NADH, exist in the cells as interconvertible members of a pool whose total concentration does not vary significantly with time. Hence, if NAD+ is oxidizing carbohydrates and lipids to produce energy in the form of ATP, levels of NAD+ would begin to fall as NADH rises. A mechanism must be present to regenerate NAD+ from NADH if oxidation continues. As we will see later, this happens in the muscle under anaerobic conditions (if dioxygen is lacking, as when you are running a 100 or 200 meter race, or if a saber-toothed tiger is chasing you) when pyruvate and NADH react to form lactate + NAD+. The reaction is shown in Figure \(\PageIndex{3}\).

Figure \(\PageIndex{3}\): Conversion of pyruvate to lactate
Under aerobic conditions (sufficient dioxygen available), NADH is reoxidized in the mitochondria by electron transport through various mobile electron carriers, which pass electrons to dioxygen (using the enzyme complex cytochrome C oxidase) to form water.
NAD+/NADH can undergo two-electron redox steps, in which a hydride is transferred from an organic molecule to the NAD+, with the electrons flowing to the positively charged nitrogen of NAD+, which serves as an electron sink. NADH does not react well with dioxygen since single electron transfers to/from NAD+/NADH produce free radical species that can not be stabilized effectively. All NAD+/NADH reactions appear to involve 2-electron hydride transfers. Figure \(\PageIndex{5}\) shows both 1 and 2 electrons to NAD+.
Figure \(\PageIndex{4}\):

Figure \(\PageIndex{4}\): 1 and 2 electrons to NAD+
FAD (or flavin mononucleotide-FMN) and its reduction product, FADH2, are derivatives of riboflavin, as shown in Figure \(\PageIndex{5}\).
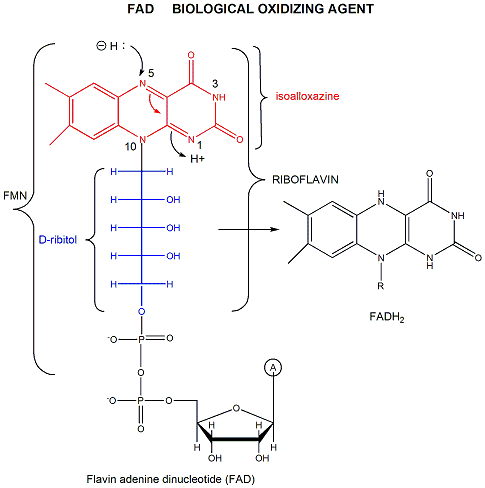
Figure \(\PageIndex{5}\): Structures of riboflavin, FMN and FAD
FAD/FADH2 differ from NAD+/NADH since they are bound tightly (KD approx 10-7 - 10-11 M) to enzymes that use them. This is because FADH2 is susceptible to reactions with dioxygen since FAD/FADH2 can form stable free radicals arising from single electron transfers. FAD/FADH2 can undergo one or two electron transfers. This is illustrated in Figure \(\PageIndex{6}\).

Figure \(\PageIndex{6}\): 1 and 2 electrons reduction of FAD
FAD/FADH2 are tightly bound to enzymes to control the nature of the oxidizing/reducing agents that interact with them. (i.e., so dioxygen in the cell won't react with them in the cytoplasm.) An enzyme with a tightly bound FAD that oxidizes a substrate would be inactive for further catalytic steps unless another oxidizing agent reoxidizes the bound FADH2.
Dehydrogenases
These enzymes use NAD+/NADH or FAD/FADH2 and are named for the oxidized substrate. For instance, in the reaction:
\[\ce{pyruvate + NADH <=> lactate + NAD^{+}} \nonumber\]
which is used to regenerate NAD+ under anerobic conditions, the enzyme is named lactate dehydrogenase. As in acid/base reactions, when the preferred direction for the reaction (from a ΔGo perspective) is from a stronger acid to a weaker (conjugate) acid, the preferred direction for a redox reaction is in the direction from a stronger to a weaker oxidizing/reducing agent. This can easily be determined from charts of standard reduction potentials and using the equation: ΔGo = -nFEo,
- where F is the Faraday constant (96,494 Coulombs/mol e- = 96, 494 J/(V.mol) = 23.06 kcal/(V.mol) or 96 kJ/(V.mol). One Faraday is the charge per one mol of electrons).
- and Eo, the standard EMF or standard cell potential (total voltage at standard state conditions), which can be determined by adding the standard reduction potentials (Eo) for the two appropriate half-reactions after reversing the equation for the half-reaction that represents the oxidation. Hence Eo=Eoreduction−Eooxidation.
When n=2 (number of electrons), which is common for oxidations of organic molecules,
ΔGo (kcal/mol) = - 46.12Eo or approximately -50E0 (or -193Eo kJ/mol)
Notice that when Eo > 0, ΔGo < 0, the reaction as written is favored under standard conditions. Note in the table below that many of the half-reactions involve protons. For biological reactions involving free protons, the standard state concentration for the protons is not 1 M as for other solutes in solution but is defined as the hydronium ion concentration at pH 7.0. The Eo and ΔGo values for the reactions involving hydrogen ions at a standard state of pH 7.0 are usually written as Eo' and ΔGo'.
Table \(\PageIndex{2}\) below shows common standard reduction potentials.
| oxidant | reductant | n (electrons) | Eo' (volts), 25oC |
|---|---|---|---|
| Acetate + carbon dioxide | pyruvate | 2 | -0.70 |
| succinate + CO2 + 2H+ | α−ketoglutarate + H2O | 2 | -0.67 |
| acetate | acetaldehyde | 2 | -0.60 |
| glycerate-3-P | glyceraldehyde-3-P + H2O | 2 | -0.55 |
| O2 | O2- | 1 | -0.45 |
| ferredoxin (ox) | ferredoxin (red) | 1 | -0.43 |
| Carbon dioxide | formate | 2 | -0.42 |
| 2H+ | H2 | 2 | -0.42 |
| α-ketoglutarate + CO2 + 2H+ | isocitrate | 2 | -0.38 |
| acetoacetate | β-hydroxybutyrate | 2 | -0.35 |
| Cystine | cysteine | 2 | -0.34 |
| Pyruvate + CO2 | malate | 2 | -0.33 |
| NAD+ + 2H+ | NADH + H+ | 2 | -0.32 |
| NADP+ + 2H+ | NADPH + H+ | 2 | -0.32 |
| FMN (enzyme bound) | FMNH2 | 2 | -0.30 |
| Lipoic acid, ox | Lipoic acid, red | 2 | -0.29 |
| 1,3 bisphosphoglycerate + 2H+ | glyceraldehyde-3-P + Pi | 2 | -0.29 |
| Glutathione, ox | Glutathione, red | 2 | -0.23 |
| FAD (free) + 2H+ | FADH2 | 2 | -0.22 |
| Acetaldehyde + 2H+ | ethanol | 2 | -0.20 |
| Pyruvate + 2H+ | lactate | 2 | -0.19 |
| Oxalacetate + 2H+ | malate | 2 | -0.17 |
| α-ketoglutarate + NH4+ | glutamate | 2 | -0.14 |
| FAD + 2H+ (bound) | FADH2 (bound) | 2 | 0.003-0.09 |
| Methylene blue, ox | Methylene blue, red | 2 | 0.01 |
| Fumarate + 2H+ | succinate | 2 | 0.03 |
| CoQ (Ubiquinone - UQ) + H+ | UQH | 1 | 0.031 |
| UQ + 2H+ | UQH2 | 2 | 0.06 |
| Dehydroascorbic acid | ascorbic acid | 2 | 0.06 |
| Ubiquinone; ox | red | 2 | 0.10 |
| Cytochrome b2; Fe3+ | Cytochrome b2; Fe2+ | 1 | 0.12 |
| Cytochrome c1; Fe3+ | Cytochrome c1; Fe2+ | 1 | 0.22 |
| Cytochrome c; Fe3+ | Cytochrome c; Fe2+ | 1 | 0.25 |
| Cytochrome a; Fe3+ | Cytochrome a; Fe2+ | 1 | 0.29 |
| 1/2 O2 + H2O | H2O2 | 2 | 0.30 |
| Cytochrome a3; Fe3+ | Cytochrome a3; Fe2+ | 1 | 0.35 |
| Ferricyanide | ferrocyanide | 2 | 0.36 |
| Cytochrome f; Fe3+ | Cytochrome f; Fe2+ | 1 | 0.37 |
| Nitrate | nitrite | 1 | 0.42 |
| Photosystem P700 | . | . | 0.43 |
| Fe3+ | Fe2+ | 1 | 0.77 |
| 1/2 O2 + 2H+ | H2O | 2 | 0.816 |
The substrate oxidation mechanism by NAD+ involves concerted hydride transfer to one face of NAD+. Hydride transfer is possible since water is excluded from the active site. It is facilitated by removing a proton from an oxygen on an alcoholic substituent, for example, adjacent to the departing hydride. The negative charge on the oxide acts as a "source" of electrons, which can then flow through the hydride transfer to the positively charged ring nitrogen of NAD+, which acts as an electron "sink." This is crudely illustrated in the cartoon shown in Figure \(\PageIndex{7}\), which shows ethanol oxidation by the enzyme alcohol dehydrogenase.

For substrates like ethanol that lose a hydride from a methylene carbon atom that has two hydrogens, only one of them is lost (either the proR or proS) from the prochiral center.
The site on NAD+ that receives the hydride and the entire ring is planar with sp2 hybridization. When bound to the enzyme, the hydride is transferred to the RE face of the ring. The same occurs in the reverse reaction when the hydride from NADH is transferred to the RE face of acetaldehyde. Using the Cahn-Ingold rules, RE and SI faces can be determined by prioritizing the substituents attached to the sp2 carbon. The reversible transfer of the proR hydrogen to the Re faces of the reactants in the reversible conversion of ethanol to acetaldehyde is shown in Figure \(\PageIndex{8}\).

Figure \(\PageIndex{8}\): Reversible transfer of the proR hydrogen to the Re faces of the reactants in the alcohol dehydrogenase reaction
FAD has a more positive reduction potential than NAD+, so it is used for more "demanding" oxidation reactions, such as the dehydrogenation of a C-C bond to form an alkene. You will notice on standard reduction potential tables that the potential of FAD is often listed several times and depends on the enzyme. This is because the FAD is tightly bound to the enzyme. Hence, its tendency to acquire electrons depends on its environment, in much the same fashion as the pKa of an amino acid side chain (which reflects its tendency to release protons), which is affected by the environment of the amino acid side chain in the protein. The standard reduction potential for flavin enzymes varies from -465 mV to + 149 mV. Compare this to the free FAD/FADH2 reduction potential, which in aqueous solution is -208 mV. The standard reduction potential of the flavin in D-amino acid oxidase, a flavoprotein, is about 0.0 V. Remember, the more positive the standard reduction potential, the more likely the reactant will be reduced and act as an oxidizing agent. Hence, the FAD in D-amino acid oxidase is a better oxidizing agent than free FAD. The KD for binding of FAD to the enzyme is 10-7 M compared to the KD for binding of FADH2, which is 10-14 M. By gaining electrons, the flavin binds more tightly, which preferentially stabilizes the bound FADH2 compared to the bound FAD. This shifts the equilibrium of FAD ↔ FADH2 to the right, making the bound FAD a stronger oxidizing agent.
A mechanism for the 2-electron hydride reduction of FAD is shown in Figure 6 above.
Figure \(\PageIndex{9}\) below shows an interactive iCn3D model of D-amino acid oxidase bound to FAD and a trifluoroalanine (1C0L) .
.png?revision=1&size=bestfit&width=618&height=330)
 Figure \(\PageIndex{9}\): D-amino acid oxidase bound to FAD and a trifluoroalanine (1C0L). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...wYfRAK9TBSt2c8
Figure \(\PageIndex{9}\): D-amino acid oxidase bound to FAD and a trifluoroalanine (1C0L). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...wYfRAK9TBSt2c8
FAD is shown in spacefill and colored yellow. Only the amino acids interacting with FAD are shown as a surface with underlying CPK-colored sticks. The noncovalent interactions are shown as sticks. If you rotate the molecule, you will see that the FAD is almost completely buried and has extensive interactions with protein, contributing to its low KD.
Thermodynamically, FAD, especially when covalently or tightly bound (and not released) from an enzyme, has a greater drive to be reduced, making it a stronger oxidizing agent than NAD+. Hence, it will be used biologically for more difficult oxidation reactions. Converting C-C to C=C is thermodynamically more difficult than converting C-OH to C=O. That can likewise be inferred from standard reduction potential (SRP) values for organic molecules. The standard reduction potential (SRP) generally varies from about +1 to +2.5V for reducing alkenes to alkanes. For reducing an aldehyde to an alcohol, the SRP generally varies from about -2.5 to about 0V.
Hence, reducing an alkene to an alkane is more favored than reducing an aldehyde to an alcohol. So, accordingly, the oxidation of an alkane to an alkene (reverse rx) is less favored than the oxidation of an alcohol to an aldehyde. This makes some intuitive sense since the alcohol is already partially oxidized. So biologically, the tough oxidations use FAD as the oxidizing agent. Of course, one of the toughest, the oxidation of H2O to O2 in photosynthesis, requires an even stronger oxidizing agent than O2 (of course). It is the oxygen-evolving complex, which we will see in the chapter on photosynthesis.
Can the standard reduction potential of a redox-active center in a protein be tuned by changing the environment of that center, much as the pKa of an acid side chain can by changing the polarity of the environment? The answer is yes. The active site of azurin, a cupredoxin, has a redox-active copper ion coordinated by a Cys and two His residues in a trigonal planar fashion. Met 121 serves as a weak axial ligand. Marshall et al. have reported a feasible method to manipulate this active site's redox potential (Eo). The wild-type azurin was mutated to alter the hydrophobicity and hydrogen bonding capabilities while maintaining the overall architecture of the metal binding site. Ser 46 was selected for mutation since it occupied a position similar to Asn in another cupredoxin involved in an important H bond binding two ligand binding loops. An N47S mutation, which strengthened the hydrogen bond between the two ligand-containing loops, increased Eo by ~130 mV while preserving metal binding site architecture as determined by UV-Vis spectroscopy. They also compared a M121Q mutant with wild-type M121 and with a M121L mutant. A plot of Eo vs. log partition coefficient for transfer of the side chain from water to octanol was essentially linear with a positive slope, showing that the standard reduction potential depended on the hydrophobicity of the weakly coordinating ligand in the metal binding region. This behavior extended to double mutants (where one set of mutants involved M121). The investigators were able to tune the Eo over a 700 mV range!
Monooxygenases
An example of monooxygenases is the hydroxylases, which hydroxylate amino acids like tryptophan and tyrosine to form 5-hydroxytryptophan and 3,4-dihydroxyphenylalanine or dopa, respectively. These two substances can be decarboxylated using PLP-dependent enzymes to form the neurotransmitters 5-hydroxytryptamine (5-HT or serotonin) and dopamine. The latter can be hydroxylated again to form norepinephrine and subsequently methylated to form epinephrine. LSD and amphetamine are analogs of serotonin and dopamine, respectively. The derivatives of tryptophan and tyrosine are shown in Figure \(\PageIndex{10}\).

Figure \(\PageIndex{10}\): Derivative of tryptophan and tyrosine derived from monooxygenases
Since these monooxygenases use dioxygen, you might expect the enzymes to use the motifs described in the previous section to facilitate their reaction with dioxygen. The enzyme contains a metal ion (Fe2+) bound to a heme in the protein. In addition, the reduction products of dioxygen that are eventually used to hydroxylate the substrate stay bound to the enzyme.
Tyrosine 3-monooxygenase (Tyrosine hydroxylase)
Tyrosine hydroxylase is a homotetramer that uses a Fe ion and biopterin cofactor for hydroxylation. Dihydroxyphenylalanine (DOPA) biosynthesis is the rate-limiting step in catecholamine synthesis.
Figure \(\PageIndex{11}\) shows a possible mechanism for the rat enzyme.

Figure \(\PageIndex{11}\): A possible mechanism for rat tyrosine hydroxylase, a monooxygenase. https://www.ebi.ac.uk/thornton-srv/m-csa/entry/134/
The Fe2+ ion binds O2. Some mechanisms show a single electron transfer to the bound O2 from the pterin ring to form Fe2+-O2-(bound superoxide) and a radical cation pterin. This is unstable and forms Fe2+-μ-peroxypterin, followed by heterolytic cleavage of the peroxo O-O bond. Ultimately, hydroxpterin and an Fe4+O oxospecies form, which then hydroxylates tyrosine.
Figure \(\PageIndex{12}\) below shows an interactive iCn3D model of rat tyrosine hydroxylase with bound cofactor analog and iron (2TOH).
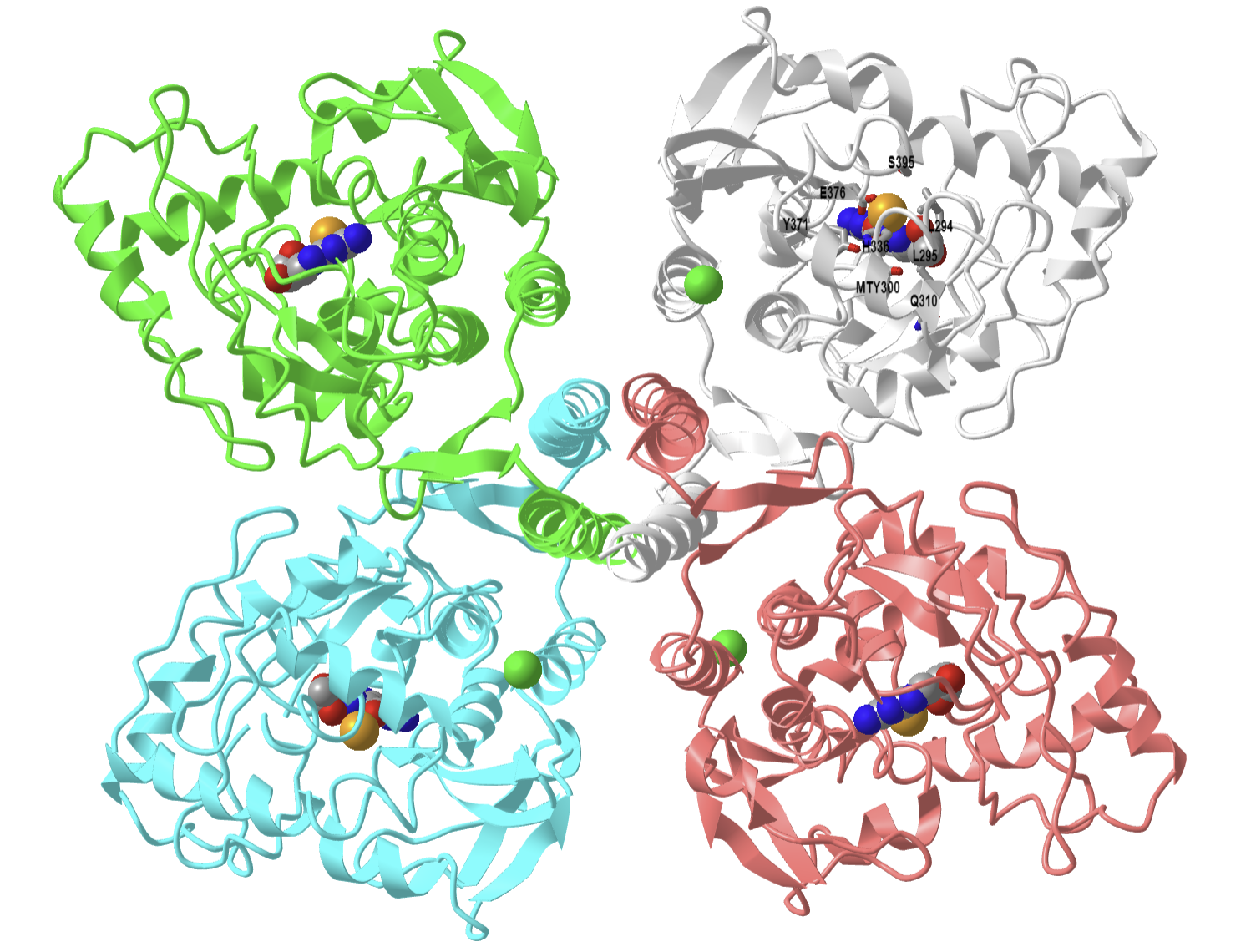
Figure \(\PageIndex{12}\): Rat tyrosine hydroxylase with bound dihydrobiopterin analogue and iron (2TOH). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...KeKiLDfmeNFfr7
Four monomers in the homotetramer are shown. The cofactor analog (HBI), 7,8-dihydrobiopterin, is shown in spacefill. Phenylalanine 300 has been self-hydroxylated by the enzyme to produce meta-tyrosine (MTY300), which, along with the other key amino acids in the active site, is labeled in CPK-colored sticks in the gray monomer. The pterin ring interacts through pi stacking with Phe 300, facilitating self-hydroxylation. The Fe ion is far (5.6 Å) from the carbon on pterin. This suggests that O2 might bridge the reacting pterin C- and the Fe ion in a Fe2+-μ-peroxypterin complex.
Tryptophan hydroxylase
This enzyme also uses a tetrahydropterin cofactor and an Fe ion to form the hydroxylating intermediates. As in the case of tyrosine hydroxylase, both the amino acid substrate and the cofactor must be present for oxygen to be activated. Otherwise, it only oxidizes Fe2+ to Fe3+. This serves as a protective mechanism so that O2 is not activated unnecessarily, which avoids the formation of soluble ROS.
Figure \(\PageIndex{13}\) shows a possible mechanism without catalytic residues for tryptophan hydroxylase.

Figure \(\PageIndex{13}\): possible mechanism without catalytic residues for tryptophan hydroxylase, adapted from Kenneth M. Roberts, Paul F. Fitzpatrick, https://doi.org/10.1002/iub.1144
Cytochrome P450s
The cytochrome P450 (CYP)consists of a large group of monooxygenases that contain a heme that absorbs maximally at 450 nm. They catalyze the following reaction:
RH + NAD(P)H + H+ + O2 → ROH + NAD(P)+ + H2O
An example includes cytochrome P450cam, which hydroxylates camphor, a large aromatic, completely nonpolar molecule. The enzyme has been called the"biological equivalent of a blowtorch," as it can, at room temperature, stereospecifically hydroxylate nonactivated hydrocarbons at physiological temperature. This reaction proceeds without stereospecificity only at high temperatures without a catalyst. Remember from the previous chapter section that dioxygen is a ground-state triplet radical that is not reactive with carbon atoms (which are singlets) unless converted to a singlet state, a process that requires significant energy. Cytochrome P450s get around this problem by binding to an Fe ion in the heme to form common intermediates such as oxos, oxides, and peroxides. The molecular orbitals of the bound dioxygen are "singlet-like."
Remember from introductory chemistry that transition metal ligands are named with a specific nomenclature. Some are shown in Figure \(\PageIndex{14}\).

Figure \(\PageIndex{14}\): Common oxygen transition metal ligands
In their names, η is the hapticity (the number of atoms of a ligand attached to a metal), and µ is the number of metal atoms bridged by a ligand. For Fe ions and oxygen ligands, some examples include FeIII–O2− (superoxo) and FeIV=O (ferryl-oxo)
If the aromatic substrate gets oxidized, something must be reduced. That something is, of course, O2. Electrons for the reduction of O2 come from the oxidized substrate and input of electrons through NAD(P)H. Cytochrome P450s are microsomal proteins, and most require another protein, NADPH-cytochrome P450 reductase (CPR). Before we look at the structure and mechanism of cytochrome P450, let's look at this microsomal protein first. CPR catalyzes this reaction:
NADPH + oxidized CytoP450 (Fe3+-heme b) + H+ ↔ NADP+ + Oxidized CytoP450 (Fe2+-heme b)
The protein contains multiple domains and results from a fusion of genes from flavodoxin (which binds FMN) and FAD reductases. In addition, it contains an NADP binding domain. It ultimately accepts a hydride ion (2 electrons) from NADPH and then transfers electrons to FMN (in one-electron steps). These are used to reduce the heme, which then activates dioxygen (oxidation number of 0) for hydroxylation reactions by cytochrome P450. In the hydroxylated organic product and water, oxygen has an oxidation number of -2.
Figure \(\PageIndex{15}\) below shows an interactive iCn3D model of rat NADPH-cytochrome P450 reductase (1AMO).
.png?revision=1&size=bestfit&width=455&height=370)
Figure \(\PageIndex{15}\): Rat NADPH-cytochrome P450 reductase (1AMO). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...nMh9ByRbsWk4B8
NADP+ is labeled as NAP. The enzyme is unique because it has binding sites for FAD and FMN (similar to nitric-oxide synthase). The flavins are aligned for electron transfer. The cleft in the enzyme presumably binds cytochrome P450. The protein has short (20 amino acids) lumenal sections, followed by a 20 amino acid membrane-spanning helix (not shown in the above structure), followed by a large cytosolic domain, where it can interact with cytochrome P450, which is found in multiple places, including the ER, microsomal, and mitochondrial membranes as well as the cytoplasm.
The cytochromes P450s contain a heme, which, instead of reversibly carrying dioxygen (as in myoglobin and hemoglobin), activates dioxygen for hydroxylation reactions involving aromatic, nonpolar substrates. Hydroxylation of these substrates increases their solubility, facilitating their elimination from the body. Epoxide intermediates and products are often produced, which can open up through nucleophilic attack using an alcohol (sugar derivative), as illustrated in Figure \(\PageIndex{16}\).

Figure \(\PageIndex{16}\): Cytochrome P450 hydroxylation reaction to increase target solubility
This type of reaction also converts steroids to different biologically active ones.
The hydroxylation reaction can also lead to reactions with amines, including those on nucleotide bases in DNA, resulting in the formation of large adducts, as illustrated in Figure \(\PageIndex{17}\).

Figure \(\PageIndex{17}\): Cytochrome P450-mediated formation of carcinogens.
Hence, cytochrome P450 can activate aromatic substrates to become carcinogens.
The cytochrome P450 family of genes/proteins is inducible on exposure to nonpolar aromatic molecules such as dioxin. These nonpolar molecules can enter the cytoplasm, binding to the arylhydrocarbon receptor (AhR), which is bound to a heat shock protein, Hsp90. Upon binding of dioxin, TCDD, for example, the AhR-TCCD complex dissociates from Hsp90 and migrates to the nucleus, where it binds a protein called Amt. The AhR-Amt complex is an enhancer/transcription factor, facilitating the transcription of the cytochrome P450 genes.
Figure \(\PageIndex{18}\) illustrates the activation of cytochrome P450 gene expression on exposure to nonpolar aromatic molecules such as dioxin.

Figure \(\PageIndex{18}\): Activation of cytochrome P450 and other gene expression on exposure to nonpolar aromatic molecules such as dioxin
On binding to a ligand, the AhR is activated and enters the nucleus, where it binds to ARNT on the aryl hydrocarbon response element (AhRE) and promotes transcription of downstream genes, including cytochrome P450 family 1 subfamily A member 1 (CYP1A1) and interleukin-1 (IL-1). CYP1A1 is in the CYP1A family and promotes the activation of procarcinogens and the hydroxylation of steroid hormones like estrogens. It also participates in the metabolism of steroidal hormones, including estrogens. ARNT is the aryl hydrocarbon receptor nuclear translocator; AhRE the aryl hydrocarbon response element; CYP1A1, XAP2 is the aryl hydrocarbon receptor interacting protein; AHRR is the aryl-hydrocarbon receptor repressor; IL-11 is interleukin 17, Hsp90 is heat shock protein 90, and p23 is prostaglandin E synthase 3
Dioxin has been shown to affect estrogen-mediated activities. Estrogens, small hydrophobic hormones derived from cholesterol, enter cells and bind to cytoplasmic estrogen receptors, which then dimerize and bind to the estrogen response element (ERE), initiating transcription. Tamoxifen, a drug derived from the yew plant, blocks the biological effects of the estrogen receptor. Although it binds to the estrogen receptor, it doesn't elicit the same conformational changes in the protein, which prevents the bound receptor from binding to the estrogen response element and recruiting other proteins needed for estrogen-dependent gene transcription. It is used in chemotherapy and the prevention of estrogen-dependent breast cancer cells.
How does dioxin interfere with estrogen signaling? Ahr and ARnt contain a basic helix-loop-helix motif, which mediates their interaction with DNA. Upon binding of the complex, detoxification genes are activated. The dioxin-Ahr-Arnt complex can also bind to the estrogen receptor, activating genes containing an estrogen response element (ERE) in the absence of estrogen. However, if estrogen is present, inhibition of gene expression from ERE is observed. Dioxins can be potent dysregulators of estrogen-induced gene expression. Such changes in estrogen activity could help to explain the pro- and inhibitory effects of dioxin on estrogen-mediated cellular responses and the possible effects of dioxin on the immune system and cancer development.
Given the importance of the cytochrome P450s, we'll offer two variant portrayals of their mechanism. Figure \(\PageIndex{19}\) shows the overall catalytic cycle of the enzyme with associated redox changes in the generic substrate, RH, and the Fe heme ion.

Figure \(\PageIndex{19}\): Catalytic cycle of cytochrome P450 with the generic substrate RH and the Fe heme ion
The key hydroxylating agent appears to be formal FeO3+, shown in step 7.
A possible mechanism for the hydroxylation of estrone by cytochrome P450 from Bacillus megaterium is shown below in Figure \(\PageIndex{20}\). It follows the general catalytic cycle shown above.
Estrone + NADH + H+ + O2 → 2-hydroxyestrone + NAD+ + H2O

Figure \(\PageIndex{20}\): Possible mechanism for the hydroxylation of estrone by cytochrome P450 from Bacillus megaterium after https://www.ebi.ac.uk/thornton-srv/m...csa/entry/699/
As with the previous generic mechanism, the two added electrons derive from NADH, which passes a hydride to FAD, which passes single electrons onto FMN, then passes them onto the heme. The exact species for several parts of the mechanism are not completely clear. For instance, a Fe4+-oxo complex has been proposed.
Let's show the structure of a human cytochrome P450 1A1 active in drug metabolism and the activation of benzo[a]pyrene (a fused benzene and α-pyrone ring), a component of cigarette smoke, into a carcinogen. Figure \(\PageIndex{21}\) below shows an interactive iCn3D model of human microsomal (ER) cytochrome P450 2A6 complexed with coumarin (1Z10). Coumarin is a benzo-α-pyrone found in many plants like tonka beans and some types of cinnamon. It has a sweet, vanilla-like scent and bitter taste.
.png?revision=1&size=bestfit&width=287&height=243)
Figure \(\PageIndex{21}\): Human microsomal cytochrome P450 2A6 complexed with coumarin (1Z10). (Copyright; author via source). Click the image for a popup or use this external link: https://www.ncbi.nlm.nih.gov/Structu...J8LxFpEyGdeab6
The heme (heme) and coumarin (COU) are shown in spacefill, CPK colors, and labeled. The amino acids lining the binding pocket for coumarin are shown in CPK-colored sticks and unlabeled, except for Asn 297. The coumarin is bound in a completely nonpolar site but forms one hydrogen bond with Asn 297 (labeled), which helps position it.
Dioxygenases
An example of a dioxygenase is the cyclooxygenase activity of prostaglandin synthase. This enzyme, often called cyclooxygenase or COX, is an integral membrane protein found in the ER membrane and a homodimer (with two hemes). It catalyzes two different reactions. One is the addition of two dioxygens to arachidonic acid - 20:4Δ5, 8, 11, 15 (which is liberated from the C2 position of phospholipid membranes by phospholipase A2 upon appropriate signaling) to form prostaglandin PGG2. This molecule, with 5 chiral centers, arises from arachidonic acid, which has one. The cyclooxygenase activity is buried in the membrane, from where the arachidonic acid can readily access its active site. The active site is at the end of a hydrophobic channel (arachidonic acid binding site) and stretches from the membrane-binding region to a buried heme. PGG2 can be further metabolized to PGH2 by the addition of two electrons to PGG2 by the hydroperoxidase activity of the enzyme, located at the other end of the enzyme. This activity forms an alcohol from the peroxide functional group in PGG2. There is one heme per monomer, which acts in both the cyclooxygenase and peroxidase activities. Each monomer of the dimer has both enzymatic activities. A possible abbreviated reaction mechanism is shown in Figure \(\PageIndex{22}\).

Figure \(\PageIndex{22}\): A possible abbreviated reaction mechanism for dioxygenase cyclooxygenase.
In summary:
- The carboxylate of arachidonic acid is coordinated to Arg 120 and Tyr 355.
- The C13 pro(S) H atom of arachidonic acid is close to Tyr 385, which allows its abstraction.
- This results in a radical centered on C11 that reacts with dioxygen to form a peroxyl radical.
- Attack by dioxygen at C11 occurs from the side of the substrate opposite to that of hydrogen abstraction.
- The oxygen radical at C11 cyclizes by attacking C9.
The C13 proS hydrogen atom (not proton) is removed from bound arachidonic acid by a free radical form of Tyr 385, which acts as an oxidizing agent. A site-specific mutant in which Phe replaces Tyr 385 is inactive. How is the Tyr free radical formed? Based on the single electron standard reduction potential (0.9 V for Tyr and -0.2 to + 0.2 V for Fe3+ in the bound heme), it appears that the heme iron is not a potent enough oxidizing agent to accomplish this task. However, oxygen bound to the heme iron could be converted to a peroxide and form an Fe4+-oxo complex (which has also been proposed for cytochrome P450). The Fe4+ ion is a more potent oxidizing agent (standard reduction potential of approximately 1 V, sufficient for oxidation of Tyr 385. Another possibility is that the peroxide activator (in forming the ferryl-oxo ligand) is NO (nitric oxide, a free radical). NO is formed by immune cells (like macrophages) on immune activation. The NO might react with superoxide (also a radical, possibly formed during an oxidative burst in macrophages during immune stimulation) to form peroxynitrite (NO3-). This can donate an oxo group to the Fe3+ to form the Fe4+-oxo complex, which could then oxidize Tyr to the free radical form. There might be other mechanisms as well to generate the Tyr free radical, since just adding organic peroxides to the enzyme will generate it.. After abstraction of the proS H atoms, a carbon-centered free radical at C11 results, which reacts with oxygen, as shown below. The exact form of oxygen that reacts is unclear, but presumably is either a peroxy or activated singlet form.
Oxidases
This class of enzymes does not incorporate dioxygen into an organic substrate. Rather, it accepts electrons released from an organic substrate through intermediate electron carriers (such as ubiquinone and cytochrome C) to form superoxide (as in NADPH-oxidase), hydrogen peroxide (as in xanthine oxidase), or water (as in cytochrome C oxidase). The mechanism of cytochrome C oxidase again supports our expectations about enzymes that use dioxygen. Dioxygen binds to metals in the enzyme. One oxygen atom binds a heme Fe2+ of cytochrome a3, which is bound to the enzyme, while the other binds a Cu1+ of Cu B. All oxygen reduction intermediates remain bound to the enzyme. Four electrons are added from four different cytochrome C molecules, which serve as mobile carriers of electrons.
We will explore cytochrome C oxidase in detail in Chapter 19.1: Electron-Transfer Reactions in Mitochondria. Figure \(\PageIndex{23}\) shows cartoon versions of several oxidases.

Figure \(\PageIndex{23}\): Examples of oxidases
Another example of an oxidase is monoamine oxidase. Mitochondrial monoamine oxidase catalyzes the oxidative deamination of certain neurotransmitters after post-synaptic neurons have taken them up in the process of inactivation. The reaction is shown in Figure \(\PageIndex{24}\).

Figure \(\PageIndex{24}\): Monoamine oxidase reaction
A Schiff base is formed, which is then hydrolyzed, incorporating unlabeled oxygen into the oxidized molecule.
Biological Oxidations: Methane to CO2
In the previous chapter section, we discussed the progressive oxidation of methane by 2 electron loses to form methanol, formaldehyde, formic acid, and CO2, with a progressive increase in oxidation number for the C by +2 (from -4 in methane to +4 in CO2), as reviewed in Figure \(\PageIndex{25}\).

Figure \(\PageIndex{25}\):: Progressive stages in the oxidation of methane
Methanotrophs are aerobic bacteria that use methane as an energy source, converting it in a series of two-electron oxidations, as shown above, to carbon dioxide. The enzymes involved in this sequential process are methane monooxygenase, methanol dehydrogenase, formaldehyde dehydrogenase, and formate dehydrogenase. Methane monooxygenase exists in a soluble and membrane form, both of which are part of a larger complex. Both have a hydroxylase (which uses dioxygen to add O to methane), and the membrane form has recently been shown to be associated with methanol dehydrogenase in a larger complex consisting of trimers of each enzyme (the hydroxylase and the dehydrogenase).
Heme Proteins
So far in this course, we have examined three different kinds of heme proteins.
- The first is that hemoglobin and myoglobin serve as carriers of dioxygen. Even though they bind one of the best oxidizing agents around (dioxygen), the heme Fe2+ is not oxidized to Fe3+. If it does, as in the case of met-Hb, the protein loses its ability to carry oxygen.
- Cytochrome C, conversely, does not bind dioxygen but serves as a carrier of electrons, which are passed to dioxygen in Cytochrome C oxidase. As an electron carrier, its Fe ion readily cycles between the 2+ and 3+ states.
- Finally, the Fe2+ in the heme of the cytochrome P450s (so named since they have an absorbance maximum at 450 nm when they bind CO) does both. It binds dioxygen and cycles between the 2+ and 3+ states, activating dioxygen for hydroxylation reactions.
The heme and amino acid ligands' structure and their absorbance spectra are shown in Table \(\PageIndex{3}\) below.
| hemoglobin | 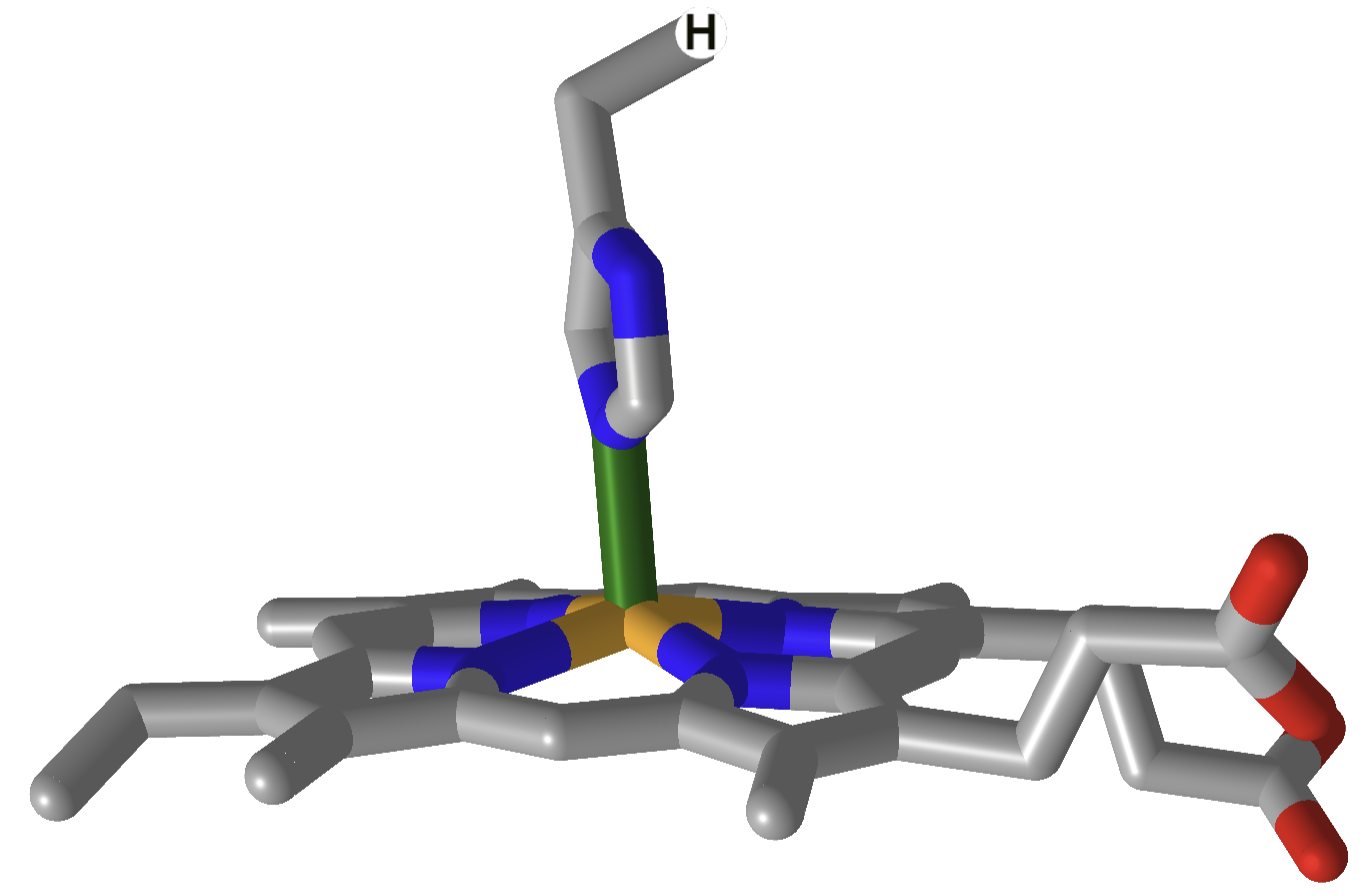 |
 |
| cytochrome C | 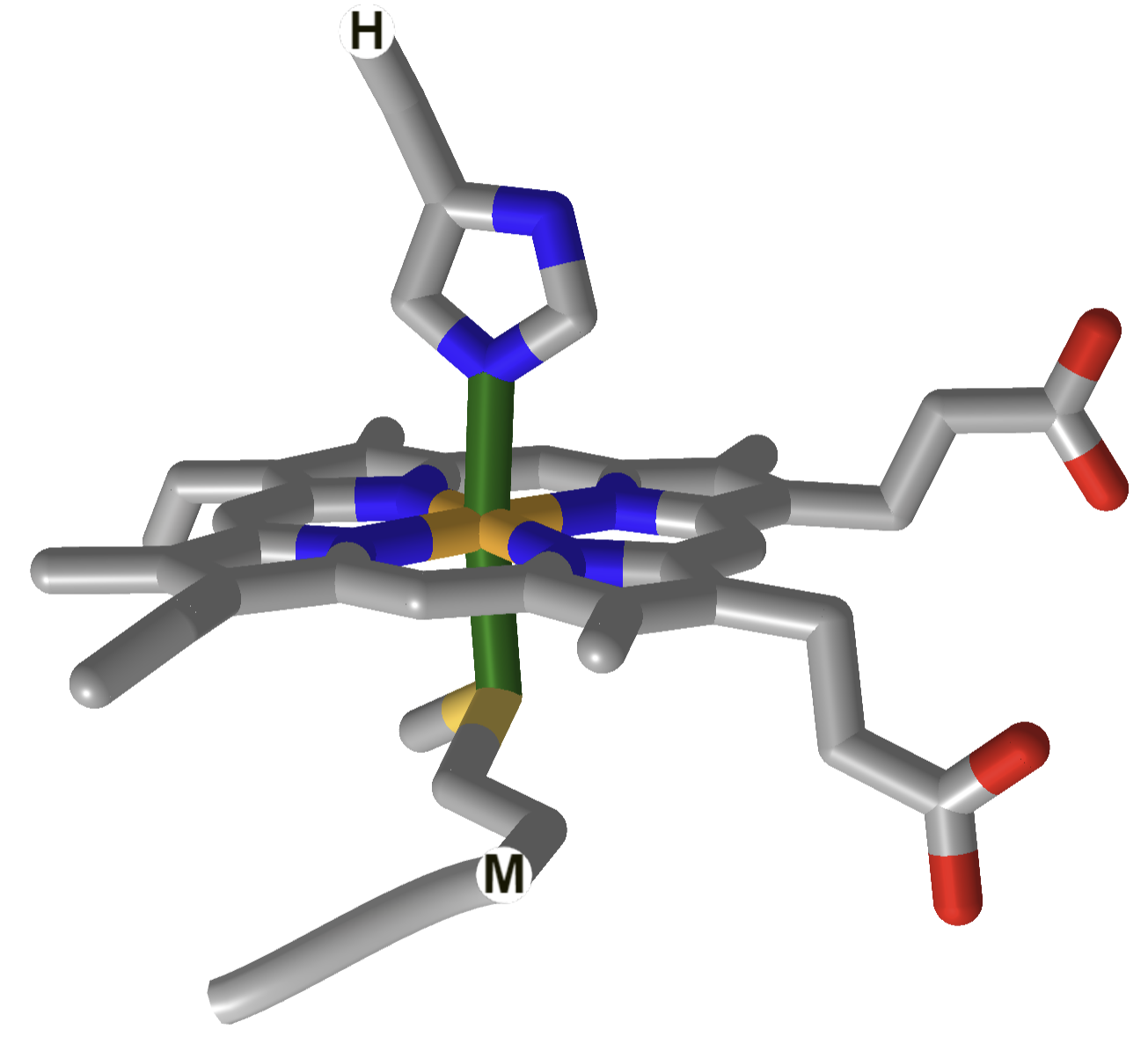 |
 |
| Cytochrome P450 | 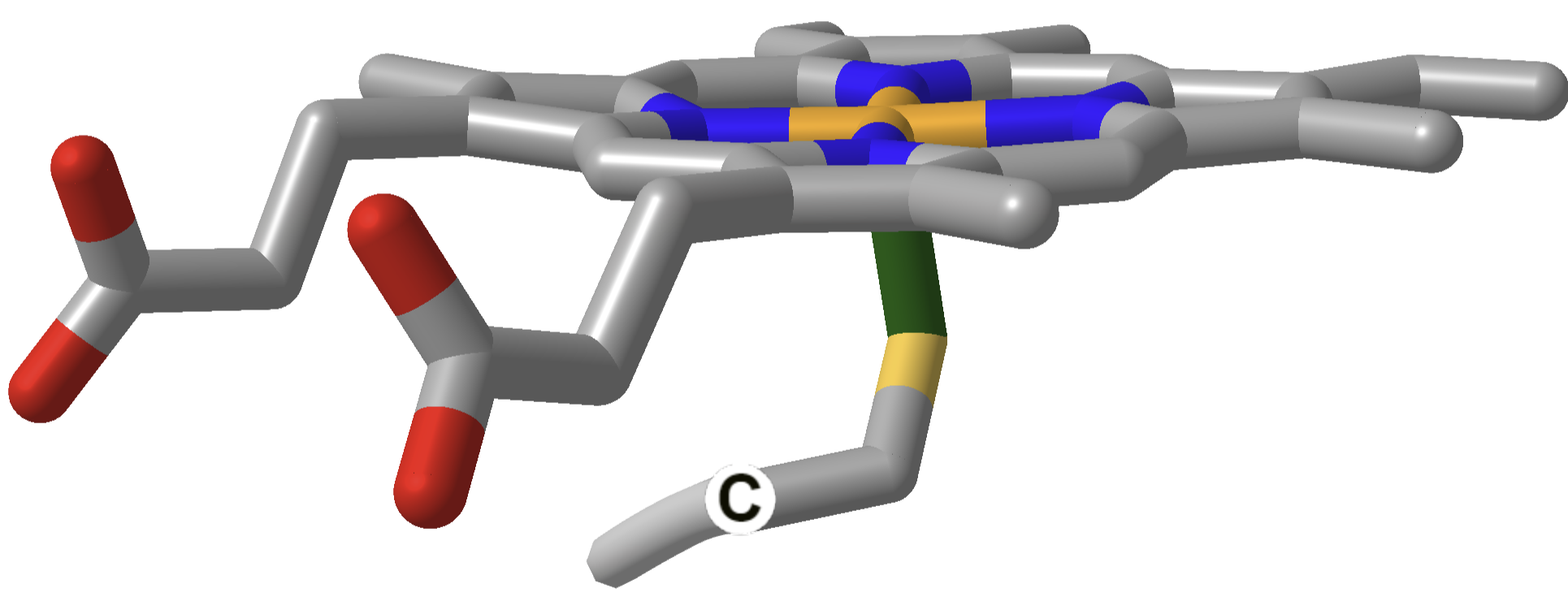 |
 |
P450: Fujishiro et al. J Biol Chem. 2011 Aug 26; 286(34): 29941–29950. Published online 2011 Jun 30. doi: 10.1074/jbc.M111.245225. CC BY license.
Cyto C: Hulko et al. December 2011. Sensors 11(6):5968-80. DOI:10.3390/s110605968. Creative Commons Attribution 3.0 Unported
Hemoglobin: Nitzan et al. July 2014, Medical Devices: Evidence and Research 7(1):231-9. DOI:10.2147/MDER.S47319. Creative Commons Attribution-NonCommercial 3.0 Unported
How could heme serve such diverse functions? We can explain this by referring to one of the course's main themes - structure mediates function. The environment of each heme must be different. The protein ligands coordinating the Fe ions are different. The 5th ligand is the proximal His in hemoglobin, while dioxygen binds to the 6th site. The 5th and 6th ligands in cytochrome C are His and Met, respectively. In cytochrome P450, the 5th site is occupied by Cys, and the 6th by dioxygen. Presumably, the environments surrounding the hemes are different as well. Once again, we have seen an analogous example in which the microenvironment influences chemical properties. The pKa of a given amino acid side chain can vary considerably depending on the polarity of the local environment. Likewise, the standard reduction potential of tightly bound FAD/FADH2 depends on the microenvironment.
As we have seen (from the study of heme proteins and the oxidative enzymes of cells), transition metals such as Fe, Zn, and Cu have vital biological roles as binding sites and cofactors in many reactions. Yet they also pose problems since they can lead to cellular oxidative damage. As we saw with cytoplasmic metallothioneins, which bind to heavy metals and protect the cell from such damage, many proteins are involved in the binding and regulating transition metals in the cell. Integral membrane proteins are required to bind and transport these cations into the cytoplasm. Other proteins act as sensors of transition ion concentration (such as latent transcription factors, which bind heavy metals and become active transcription factors for metallothioneins. Others act as chaperone proteins, which bind metal ions and transfer them to apometalloproteins. Recent work suggested transporters and chaperones are involved in metal ion biology and bind these ions with unusual coordination geometry, presumably facilitating the transfer of the ion to the apo-target protein.
The transition metals Zn and Fe are often found in E. Coli at a concentration of 0.1 mM, compared to Cu and Mn, which are present at 10 to 100 μM concentrations. Also, about one-third of all proteins demonstrate specific binding of metal ions and can be classified as metalloproteins. Mass balance suggests that metal ions would be distributed in proteins with low, intermediate, and high metal binding affinity as well as in free pools, which potentially can be toxic to cells. Depending on their Kd for metal ion binding, metalloproteins would be in various states of ligation. The free concentration of some ions (Cu and Zn) is so low that newly synthesized apoproteins that bind these ions would not obtain the ions from the free pool. In such cases, metal chaperones would be required.
Summary
This chapter provides an integrated overview of the principles and mechanisms underlying oxidation reactions in both chemical and biological contexts. It bridges foundational organic redox chemistry with the sophisticated enzyme-catalyzed processes essential for metabolism and detoxification in living systems.
-
Chemical Oxidation Reactions:
The chapter begins by reviewing classical oxidation mechanisms encountered in organic chemistry. It outlines the multi-step process of oxidation—highlighting initiation, propagation (including branching), and termination steps—using the combustion of methane as an illustrative example. In laboratory settings, strong oxidants like permanganate and chromate are employed to achieve controlled oxidation, contrasting sharply with the challenges of using dioxygen, whose uncontrolled reactivity and complex radical intermediates often lead to overoxidation and side reactions. -
Biological Oxidizing Agents:
In biological systems, oxidation is precisely regulated. Rather than relying on dioxygen directly for substrate oxidation, cells use specialized redox carriers such as NAD⁺ and FAD. These cofactors mediate two-electron hydride transfers via dehydrogenase enzymes, which are central to energy-yielding pathways like glycolysis, the Krebs cycle, and fatty acid oxidation. The chapter explains the structural basis and mechanistic details of hydride transfer, emphasizing how the planar, electron-deficient nicotinamide ring of NAD⁺ acts as an effective electron sink. -
Enzyme Classes Utilizing Dioxygen:
The discussion expands to enzymes that use molecular oxygen in more diverse ways:-
Monooxygenases (Hydroxylases):
These enzymes incorporate one atom of dioxygen into a substrate while reducing the other to water. Examples include tyrosine and tryptophan hydroxylases, which are critical in neurotransmitter biosynthesis. -
Dioxygenases:
Unlike monooxygenases, dioxygenases insert both atoms of dioxygen into their substrates. The cyclooxygenase activity of prostaglandin synthase, which converts arachidonic acid to prostaglandin precursors, is presented as a key example. -
Oxidases:
Oxidases catalyze reactions in which electrons from substrates are transferred to dioxygen, producing reactive oxygen species (ROS) such as superoxide or hydrogen peroxide. The chapter outlines how enzymes like cytochrome C oxidase and monoamine oxidase harness oxidase activity to facilitate vital cellular processes, including terminal electron transfer and neurotransmitter inactivation.
-
-
Cytochrome P450 and Electron Transfer Mechanisms:
A substantial portion of the chapter is devoted to cytochrome P450 enzymes, the workhorses of xenobiotic metabolism. These heme-containing monooxygenases use electrons supplied by NADPH-cytochrome P450 reductase to activate dioxygen, enabling the hydroxylation of nonpolar, otherwise inert substrates. The catalytic cycle of P450s, with its series of redox intermediates and oxygen-activating steps, is examined in detail, along with discussions of how structural variations in the heme environment govern function. -
Tuning Redox Potentials and Transition Metal Homeostasis:
The chapter highlights the principle that an enzyme’s microenvironment can tune the standard reduction potentials of its redox-active centers. Examples include modifications in FAD-binding sites and engineered mutations in cupredoxins (e.g., azurin) that modulate redox potentials by altering hydrophobicity and hydrogen bonding. Additionally, the role of metalloproteins in managing transition metal ions (Fe, Zn, Cu) is discussed. These proteins not only support essential catalytic functions but also prevent metal-mediated oxidative damage by sequestering free ions through high-affinity binding and specialized chaperone systems. -
Integration into Metabolism and Physiology:
Finally, the chapter connects these oxidation mechanisms to the broader context of cellular metabolism. The sequential oxidation of methane by methanotrophs exemplifies how a series of enzyme-catalyzed redox reactions can fully extract energy from an organic substrate. Likewise, the diverse functions of heme proteins—from oxygen transport by hemoglobin to electron transfer by cytochrome C and substrate oxidation by cytochrome P450—underscore the versatility of redox chemistry in biology.
In summary, this chapter underscores how nature has evolved a variety of oxidizing agents and enzymes to precisely control redox reactions. These processes are fundamental not only for energy production and biosynthesis but also for detoxification and metabolic regulation. Understanding these mechanisms is crucial for appreciating how redox chemistry underpins life at the molecular level.