
6.1: How Enzymes Work
- Page ID
- 102264


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)(Learning goals written by Claude, Sonnet 4.6, Anthropic)
Chemical Strategies for Rate Enhancement
- Distinguish specific acid/base catalysis (rate depends solely on [H₃O⁺] or [OH⁻]) from general acid/base catalysis (rate depends on buffer component concentration), explain how general acids donate protons to reduce developing negative charge in the transition state and general bases abstract protons to reduce developing positive charge, and identify the amino acid side chains (Asp, Glu as general acids; His imidazole as both general acid and base) that serve these roles in enzyme active sites.
- Explain metal ion (electrostatic) catalysis — including how Lewis acid metals like Cu²⁺ and Zn²⁺ stabilize oxyanion intermediates in decarboxylation reactions, how Zn²⁺ in carbonic anhydrase reduces the pKa of a coordinated water molecule from 14 to ~9, generating a hydroxide nucleophile at physiological pH — and connect the prevalence of this mechanism to the observation that approximately one-third of all enzymes require metal ion cofactors.
- Explain covalent (nucleophilic) catalysis — in which a nucleophilic catalyst (such as pyridine or an enzyme active site residue) forms a transient covalent intermediate with the substrate, splitting the original high-energy transition state into two lower-energy ones — and describe the role of iminium ions (Schiff bases) as powerful electron sinks that promote C–C and C–H bond cleavage, using Schiff base formation between a lysine residue and a keto-substrate (fructose bisphosphate aldolase) as a biochemical example.
Transition State Stabilization and the Thermodynamic Basis of Catalysis
- Use the linked equilibrium diagram (Pauling's formulation) to derive mathematically that an enzyme must bind the transition state (S‡) more tightly than the substrate (S) to catalyze a reaction — expressing this as KT > KS and kE/kN = KT/KS — and calculate that if KT/KS ~ 10⁸–10¹⁴ and KS ~ 10³–10⁵ M⁻¹, the enzyme binds S‡ with KD ~ 1 femtomolar, comparable to the avidin-biotin interaction.
- Explain the abzyme (catalytic antibody) concept as experimental proof of Pauling's transition state stabilization hypothesis — describing how immunizing mice with a stable phosphonate transition state analog of an ester elicits antibodies that catalyze ester hydrolysis — and explain why transition state analogs are used as tight-binding enzyme inhibitors in drug design.
Physical Strategies for Rate Enhancement
- Quantify the rate advantage of intramolecular over intermolecular reactions using the ratio k₁/k₂ — which has units of effective molarity and can reach 10⁵–10⁸ M for sterically restricted systems — explain this advantage in terms of the loss of translational and rotational entropy in bimolecular reactions (ΔS ~ −35 cal/K·mol in solution, equivalent to an effective concentration of 10⁸–10⁹ M), and connect this to enzyme catalysis where substrate binding converts the subsequent chemical step into an effectively intramolecular reaction.
- Explain strain distortion as a physical catalytic mechanism — in which induced fit conformational changes in the enzyme active site upon substrate binding destabilize the substrate toward the transition state geometry or impose unfavorable bond angles that lower the activation energy — and distinguish this from purely chemical catalytic strategies.
- Describe asymmetric (organocatalysis) as an extension of enzyme catalytic principles to small molecules — noting that L-Pro can form a Schiff base intermediate to catalyze asymmetric aldol condensations producing a single enantiomer, analogous to enantiospecific enzyme active sites — and explain why asymmetric catalysis (recognized by the 2021 Nobel Prize in Chemistry for List and MacMillan) matters for pharmaceutical synthesis.
In this section, we will explore chemical and physical factors that accelerate reactions and begin to relate these effects to those of enzymes. We will see that enzymes employ various chemical strategies to increase reaction rates, in addition to physical strategies such as reactant proximity and the introduction of strain. This can result in reactions 10 million (or more!) times faster than the uncatalyzed reaction. To put this 10-million-fold rate enhancement in perspective, if the catalyzed reaction takes 1 second, the uncatalyzed one would take nearly 4 months!
This chapter section has been written by Kristen Procko and Henry Jakubowski.
Reactions in uncatalyzed solutions are slow. Consider the hydrolysis of an ester in water, illustrated in Figure \(\PageIndex{1}\). The ester is stabilized by resonance and is therefore weakly electrophilic; thus, attack by weakly nucleophilic water is slow. Examining the transition state, we see that charge development and separation occur in the transition state for the uncatalyzed reaction, resulting in an intermediate (P) with both positive and negative charges.

Figure \(\PageIndex{1}\): Charge development in the transition state during ester hydrolysis
When bonds are made or broken, charged intermediates are often formed, which are higher in energy than the reactants. Consider the energy diagram for the first step of a generic endergonic reaction, shown in \(\PageIndex{2}\). The transition state is closer in energy to the intermediate P than to the reactant R. Therefore, the TS more closely resembles P than the starting reactants. Applying this analysis to the ester hydrolysis reaction from Figure \(\PageIndex{1}\), the transition state is closer in energy to the charge-separated intermediate P and, therefore, more closely resembles the charge-separated species. In this example, the intermediate is higher in energy than the reactants; thus, the transition state is even higher in energy than the intermediate.

Figure \(\PageIndex{2}\): An energy diagram for an endergonic reaction (Image modified from: Chemistry LibreTexts, 6.9)
Anything that can stabilize the charges on the intermediate will also stabilize the developing charges in the transition state. This lowers the energy of the transition state, thereby catalyzing the reaction. This section will investigate the mechanisms underlying the catalysis of chemical reactions by small molecules. Presumably, biological macromolecular catalysts (such as protein enzymes) will use similar mechanisms in their catalysis (which will be discussed in the next section).
Catalysts, including enzymes, can stabilize transition states in at least five ways.
Chemical Strategies for Rate Enhancement
General Acid and Base Catalysis
Considering intermediate P in Figure \(\PageIndex{1}\), we can envision two strategies to reduce the charge separation: the negative charge on the anionic oxygen could be protonated, or the positive charge on the cationic oxygen could be removed by deprotonation. If the reaction is pH-dependent and the reaction rate depends solely on hydronium ion concentration ([H3O+]), then specific acid catalysis is operative. Specific acid catalysis occurs when the hydronium ion concentration is the sole factor determining the reaction rate, and the concentration of any buffer components present in the solution does not influence the rate. In other words, the reaction rate depends specifically on the concentration of hydronium ions. Specific base catalysis occurs when the reaction rate depends solely on the hydroxide ion concentration and is again independent of any buffer components in the solution.
By contrast, general acid catalysis occurs when the reaction is not solely dependent on [H3O+] concentration; that is, the concentration of a buffer component influences the reaction rate. With general acid catalysis, the charge separation in the transition state is decreased by the donation of a proton to the carbonyl from general acids (e.g., acetic acid or a protonated imidazole ring). Proton donation decreases the developing negative charge in the transition state. In Figure \(\PageIndex{3}\), the first step of the ester hydrolysis mechanism is shown via specific acid catalysis alongside the general acid catalysis mechanism using the weak acid, acetic acid.

Figure \(\PageIndex{3}\): Specific vs. general acid catalysis
Alternatively, the first step of the ester hydrolysis mechanism can be base-catalyzed, increasing the nucleophile's strength. In Figure \(\PageIndex{1}\), the attacking water molecule develops a partial positive charge in the transition state as it begins to form a bond with the electrophilic carbon of the carbonyl. In the base-catalyzed mechanism shown in Figure \(\PageIndex{4}\), hydroxide becomes the nucleophile in the specific base-catalyzed mechanism. The energy of the transition state can also be lowered by the presence of a general base (e.g., acetate, a deprotonated imidazole ring). Proton abstraction decreases the developing positive charge.

Figure \(\PageIndex{4}\): Specific vs. general base catalysis
General acid/base catalysis is common in enzymes because they often use amino acid side chains to promote acid-base reactions within the active site, the region of the enzyme where the chemical reaction occurs. Acetic acid is similar to glutamic and aspartic acid side chains, and the imidazole ring shown in the general base catalysis reaction in Figure \(\PageIndex{4}\) is present in the side chain of the amino acid histidine.
Metal Ion or Electrostatic Catalysis
A metal such as Cu2+ or Zn2+ can also stabilize the transition state. The metal must be able to bind the charged intermediate and, by extension, the transition state. An oxyanion intermediate formed during the reaction of an electrophilic carbonyl C can interact with a metal, especially when there is an O on an adjacent atom, which can help coordinate the metal ion. This charge stabilization of the developing negative charge in the transition state and the fully negative charge in the intermediate is often called electrostatic catalysis. It is illustrated in the decarboxylation of a β-keto carboxylic acid, Figure \(\PageIndex{5}\). Coordination of Cu2+ to the β-keto carboxylic acid increases the electrophilicity of the carbonyl, making it a superior electron acceptor, which facilitates decarboxylation. The intermediate enolate formed during decarboxylation is protonated, yielding the more stable ketone as the product.

Figure \(\PageIndex{5}\): Metal ion catalysis
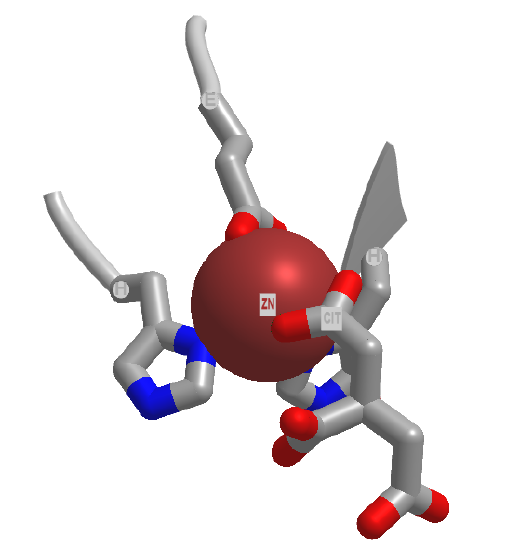
Electrostatic catalysis is likely to occur in many enzymes since nearly 1/3 of all enzymes require metal ions. A classic example of an enzyme using metal ion catalysis is carboxypeptidase A. Figure \(\PageIndex{6}\)s shows an interactive iCn3D model of Zn and the inhibitor citric acid bound to carboxypeptidase A (3KGQ). Note the histidine and aspartate amino acid side chains of the active site coordinating to the Zn2+ ion, along with the carboxylate group of citrate.
Metals can also act differently. They may coordinate a water molecule and, by further polarizing the H-O bond, increase the acidity of the bound water. For instance, a water molecule in the hexaaquairon(III) ion has a pKa of 9.4, compared to pure water, with a pKa of 14 (\(\PageIndex{7}\)). The complexed hydroxide is a better nucleophile than bulk water.

Figure \(\PageIndex{7}\): Metal ion decrease of pKa of coordinated water
Another enzyme that utilizes Zn2+ is carbonic anhydrase. It is among the fastest enzymes, with a kcat of 106 s-1 and a kcat/Km of 8.3 x 107 M-1s-1 (reference). It is diffusion-controlled at low substrate (CO2) concentration and converts one million bound CO2 per second to HCO3-! The Zn2+ appears to bind a water molecule and reduce its pKa such that the bound form is OH-. This is illustrated in Figure \(\PageIndex{8}\), which depicts the local environment of the bound Zn2+ (coordinated by three histidine side chains and an OH-) in the absence (left) and presence (right) of CO2.

Covalent or Nucleophilic Catalysis
One way to change the activation energy of a reaction is to alter the reaction mechanism, introducing new steps with lower activation energies. As shown in Figure \(\PageIndex{9}\), the catalyzed reaction has a new lower-energy well, representing the formation of the covalent intermediate, and the activation energy is lowered overall. Formation of the new intermediate results in two transition states, represented by the two high-energy points of the blue line in the plot.
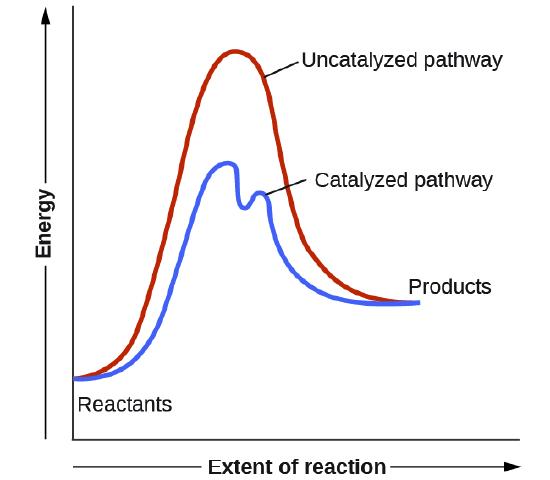
Figure \(\PageIndex{9}\): Energy diagram for an uncatalyzed reaction compared to a catalyzed reaction that utilizes covalent catalysis.
A typical way to achieve covalent catalysis is to add a nucleophilic catalyst, which forms a covalent intermediate with the reactant. Figure \(\PageIndex{8}\) shows how pyridine (red) acts as a nucleophilic or covalent catalyst in the hydrolysis of an anhydride. The anhydride is very reactive initially, and the charged pyridinium ion intermediate contains a very good leaving group. The desired nucleophile, water, can then interact with the intermediate in a nucleophilic substitution reaction. In these reactions, in general, as long as the nucleophilic catalyst is a better nucleophile than the ultimate nucleophile (usually water), the activation energy is lowered, and the reaction is catalyzed. The nucleophilic catalyst and the original nucleophile usually interact with a carbonyl C in a substitution reaction.

Figure \(\PageIndex{10}\): Nucleophilic covalent catalysis by pyridine
Reactions involving iminium ions are a recurring theme in biochemistry
Positively charged nitrogen cations (iminium ions) form as intermediates in many biochemical mechanisms. The iminium ion is a powerful electron acceptor and can promote the cleavage of bonds that would otherwise be difficult to break, such as C–C and C–H bonds. To begin our analysis of how iminium ions promote such cleavage reactions, let's revisit a common carbon-carbon bond-cleaving reaction, the decarboxylation of a β-keto acid, which we examined briefly above in Figure \(\PageIndex{5}\) with metal ion catalysis.
Under acidic conditions, β-keto acids usually decarboxylate with gentle warming. A cyclic transition state is often invoked, and the presence of the carbonyl of the ketone adjacent to the breaking bond gives the electrons somewhere to go (Figure \(\PageIndex{11}\)). The decarboxylation product is an enol, which tautomerizes to the more stable ketone. The equilibrium favors the deprotonated carboxylate form under the slightly basic conditions that characterize most biochemical reaction media. The adjacent carbonyl again gives the electrons somewhere to go in the decarboxylation reaction, and under these basic conditions, an enolate is formed. Protonation gives the final product, a ketone.

Figure \(\PageIndex{11}\): Decarboxylation of a β-keto acid under acidic conditions, and at pH 7.4
In Figure \(\PageIndex{5}\), we saw that a metal ion can promote the decarboxylation reaction by interacting with the electron-accepting ketone carbonyl, which makes it even more electrophilic. Another strategy to create a better electron acceptor involves forming a full positive charge on the electron-accepting atom, which can be done by converting the ketone to an iminium ion. Amines react with aldehydes or ketones to form iminium ions. Figure \(\PageIndex{12}\) illustrates this strategy involving covalent catalysis. The amine, RNH2, reacts to form a new intermediate, the iminium ion, with a full positive charge. The protonated nitrogen serves as an excellent electron "sink" for decarboxylation reactions of beta-keto acids.
This iminium ion or protonated Schiff Base has a pKa of about 7, so the protonated iminium and deprotonated imine are in equilibrium near pH 7. Figure \(\PageIndex{12}\) also illustrates a simple way to view reaction mechanisms. Electrons in chemical reactions can be viewed as flowing from a source (such as a carboxyl group) to a sink (such as an electrophilic carbonyl O or a positively charged N in a Schiff base).

Acid- and base-catalyzed reaction mechanisms for Schiff base formation are shown in Figure \(\PageIndex{13}\). An amine is used as the nucleophilic catalyst, forming the initial addition product, a carbinolamine. The carbinolamine dehydrates because the free pair of electrons on the N is more likely to be shared with the carbon to form a double bond than those on the original carbonyl O, which is more electronegative than the N. If catalyzed by a general acid, an iminium ion and the base-catalyzed reaction form an imine. Near pH 7.4, the imine is readily protonated, forming a positively charged N at the former carbonyl oxygen.

Figure \(\PageIndex{13}\): Schiff Base Formation - reaction mechanism
An actual Schiff base intermediate between fructose-1,6-bisphosphate (2FP-400) and Lys 239 from the enzyme fructose bisphosphate aldolase from Leishmania mexicana is shown in Figure \(\PageIndex{14}\). Only a single bond between the carbon and nitrogen in the Schiff base is shown.
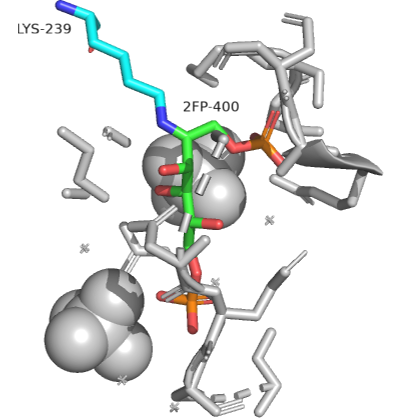
Figure \(\PageIndex{14}\): Schiff base intermediate between fructose-1,6-bisphosphate (2FP-400) and Lys 239 from the enzyme fructose bisphosphate aldolase from Leishmania mexicana(2QDG)
Transition State Stabilization
In the middle of the 20th century, Linus Pauling postulated that the only thing that a catalyst must do is bind the transition state more tightly than the substrate. This can be discerned in Figure \(\PageIndex{15}\) and a little math. The diagram shows how the substrate (S) and the transition state (S*) can react with an enzyme (E) to form a complex, which then proceeds to product (following the diagram from "start here" in the right-hand direction), or can go to product in the absence of enzyme (E) (following the diagram from "start here" in the left-hand direction). Note that the diagram is arbitrarily drawn such that the standard Gibbs free energy (G°) of the free product P is higher than that of the free substrate, S.

Figure \(\PageIndex{15}\): Enzymes bind the transition state more tightly than the substrate
The large colored vertical arrows represent the ΔG° for the transition shown:
- The red arrows A and B represent the ΔG°s for the binding of E + S (arrow A) and E + S* (arrow B), respectively.
- The green arrows C and D represent the ΔG°s for the activation energy of the free substrate (arrow C) and the enzyme-bound substrate (arrow D).
Now consider the two pairs of arrows (A, C and D, B). Add each set like a vector in elementary physics. Since the distance between the two horizontal blue lines is the same for the left-hand process (uncatalyzed) and the right-hand one (catalyzed), it follows that
\begin{equation}
C-A=D-B
\end{equation}
The negative signs for A and B are used since, in the diagram, both A and B have negative ΔG° values.
Now, for an enzyme to be a catalyst, the activation energy D for the reaction in the presence of the enzyme E must be less positive (i.e., smaller) than the activation energy C in the absence of the enzyme. Therefore, after rearranging the equation:
\begin{equation}
C-D=A-B>0
\end{equation}
and substituting in the ΔG° values for A and B, we can directly compare the free energy of binding the substrate (S) vs. binding the transition state (S*):
\begin{equation}
-R T \ln \mathrm{K}_{\mathrm{eq} \mathrm{S}}-\left(-\mathrm{RT} \ln \mathrm{K}_{\mathrm{eq} \mathrm{S}^{*}}\right)>0
\end{equation}
Hence, the equilibrium constant for binding the transition state is larger than that for binding free substrate:
\begin{equation}
\mathrm{K}_{\text {eq } S^{*}}>\mathrm{K}_{\text {eq } \mathrm{S}}
\end{equation}
Pauling was right. The enzyme just needs to bind the transition state more tightly than the substrate to catalyze the reaction. This is why chemists synthesize stable transition state analogs as potential tight-binding inhibitors of target proteins.
The stability of the transition state also affects reaction kinetics (which makes sense, given that the activation energy clearly affects reaction rate). As you probably remember from organic chemistry, biomolecular nucleophilic substitution (SN2) reactions are slow when the central atom where the substitution will occur is surrounded by bulky substituents (sterics once again). We discussed this in the context of nucleophilic substitution on a sp2-hybridized carbonyl carbon in carboxylic acid derivatives versus on a sp3-hybridized phosphorus in phosphoesters and diesters. The explanation for this phenomenon has usually been attributed to hindered access to the central atom caused by bulky substituents (intrinsic effects). Is this true? Studies on SN2 reactions of methylchloroacetonitrile and t-butylchloroacetonitrile (with the reagent labeled with 35Cl) using 37Cl- as the incoming nucleophile in the gas phase showed that the more hindered t-butyl derivative's activation energy was only 1.6 kcal/mol (6.7 kJ/mol) higher than the methyl derivative, but in aqueous solution, the difference is much greater for comparable reactions (Figure \(\PageIndex{16}\):).

Figure \(\PageIndex{16}\): SN2 reactions are characterized by a pentavalent transition state
They attributed the differences to the solvation effects of the transition state. The bulkier the substituents on the central atom, the more difficult it is to solvate the transition state since water also can't reorient around it as well. In effect, there is steric hindrance to both the reactant and the solvent.
What does it take for a macromolecule (M) to be a catalyst - an enzyme? It seems the minimum criteria are:
- M binds a reactant
- M binds the transition state more tightly than the substrate
Anything above these is just "icing on the cake." If different functional groups are present in the "active" site of the enzyme that would allow electrostatic, intramolecular, covalent, general acid and/or base catalysis, the better the catalyst.
A transition state analog case study: Abyzmes (Antibody Catalysis)
Recall that antibodies are immune system proteins that bind foreign molecules (see Chapter 5.4). The usual role of an antibody is to initiate an immune response. When the antigen-binding site, located in the variable region of an antibody, binds to an antigen, it triggers the production of new antibodies (within B cells) to optimize the immune response to that antigen. These new antibodies are made with mutations in the antigen-binding region. Those that bind more tightly than the original antibody will be selected to form longer-lived memory B cells, ready for the next time the body encounters the antigen.
Decades ago, Linus Pauling hypothesized that antibodies could be produced with an atypical role—to act as catalysts! If antibody molecules could be engineered to bind a compound resembling the transition state of a chemical reaction, they should presumably catalyze the reaction. In 1987, his prediction was verified. Lerner et al. made a transition-state analog of an ester. When an ester is hydrolyzed, as shown in Figure \(\PageIndex{17}\), the sp2-hybridized carbonyl carbon is converted to an sp3-hybridized center in the intermediate, with the carbonyl oxygen becoming an oxyanion.
The transition state presumably resembles this unstable intermediate (sp3, oxyanion). Thus, Lerner synthesized a phosphonate, an ester mimic with a sp3 hybridized phosphorous replacing the carbonyl C. It also has a negatively charged oxygen, as does the intermediate for the ester. This phosphonate ester is very resistant to hydrolysis. When injected into a mouse (after first being covalently attached to a carrier protein to make the small molecule "immunogenic"), the mouse produces an antibody that binds to the phosphonate. When the corresponding carboxylic acid ester is added to the antibody, it is cleaved with nominal kcat and KM values. Site-directed mutagenesis can then be done to make it an even better catalyst! The antibody enzymes have been called abzymes. The structure shown in Figure \(\PageIndex{17}\) also shows how phosphonamides act as transition state analogs.

Figure \(\PageIndex{17}\): PHOSPHONAMIDES: TRANSITION STATE ANALOGS
Figure \(\PageIndex{18}\)s shows an interactive iCn3D model of the transition state analog 5-(para-nitrophenyl phosphonate)-pentanoic acid bound to a mouse Fab antibody fragment with esterase activity (1aj7)
.png?revision=1&size=bestfit&width=269&height=251)
Transition state theory can be used to quantify the relationships described in the graphical analysis above. This analysis will use the equilibrium constant (in contrast to the last two chapters, which used dissociation constants to characterize ligand-binding macromolecules, receptors, and enzymes). Let's assume that a substrate S is in equilibrium with its transition state S‡. Hence Keq = [S‡]/[S]. The following reaction can be written: S → S‡ → P. Based on our previous kinetic analysis and experience in writing differential equations, dP/dt = k1[S‡]. By analogy, enzyme-bound S (ES) can be converted to (ES‡) and then on to product as shown in the following chemical equation:
\[\ce{E + S <=> ES -> ES^{†} -> E + P}. \nonumber \]
For the non-enzyme catalyzed reaction, transition state theory can be used to show that the first order rate constant k1= kT/h where k is the Boltzmann constant, T is the Kelvin temperature, and h is Planck's constant. Hence, using Keq = [S†]/[S], equation 1 can be derived
\begin{equation}
\frac{d P}{d t}=\frac{k T}{h}\left[S^{\dagger}\right]=\frac{k T}{h} K^{\dagger}[S]=k_{n}[S]
\end{equation}
where kn (hereafter written as kN) =(kT/h)K† is the effective first-order rate for the non-catalyzed rate. Now, let's create a more complicated linked equilibrium that shows the same reaction in the presence of an enzyme. Figure \(\PageIndex{19}\)

Remember that the K values for this analysis are equilibrium constants, not dissociation constants. Note two important equilibrium constants, KS, the equilibrium constant for the binding of free S to E, and KT, the equilibrium constant for the binding of free S† to E (assuming that free S† could bind to E before it converted to product). As we have seen for linked equilibrium before, since the Keq values are related to the standard free energy changes, which are state functions, the sum of the standard free energies going from E + S to ES† (by either the top or bottom paths) is path independent, so the products of the Keq for the top path are equal to those for the bottom paths. This gives the following equation:
\begin{equation}
\frac{K_{T}}{K_{S}}=\frac{K_{E^{\dagger}}}{K_{N^{\dagger}}}=\frac{k_{E}}{k_{N}}
\end{equation}
The right-hand side is the ratio of the effective first-order rate constant for conversion of ES† → E + P, kE divided by the rate constant for the conversion of S† → P for the noncatalyzed rate, kN. The final ratio of rate constants can be derived from the simple relationship that kx=(kT/h)K†x where x is either N (non-catalyzed) or E (enzyme-catalyzed). Equation 2 states that the equilibrium constant for the binding of S† to E, KT, is greater than that for the binding of S to E, KS (as kE > kN). KT/KR ranges from 108 - 1014. Given common values for the equilibrium constant for binding of S to E (103 – 105 M-1), which is equivalent to the dissociation constant values Kd = 10 uM – 1 mM, the calculated value of KT = 1015 M-1, which gives a dissociation constant for the enzyme and transition state of Kd = 10-15 M (1 femtomolar). This is as tight as one of the highest-affinity binding interactions in the biological world: the binding of avidin and biotin. As we noted in Chapter 5.1, assuming that the second-order rate constant for avidin/biotin binding and as shown above for E/S† is diffusion-controlled (about 108 M-1s-1), the off rate for the avidin-biotin or ES† complex is 10-7 s-1, equivalent to a half-life of the complex of 80 days. It doesn't get much tighter than that.
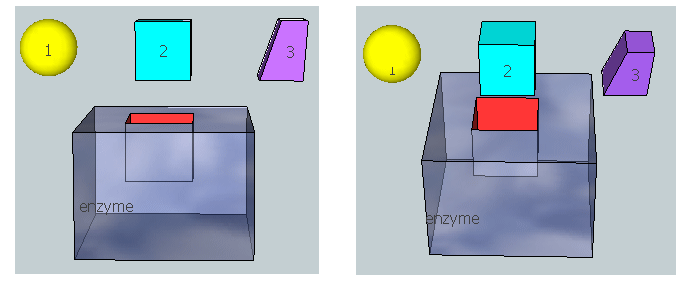
Figure \(\PageIndex{20}\) represents an image of an enzyme and three molecules, 1–3, that could bind to it. Using the analysis above, which molecule do you think represents the substrate? Transition state? Product?
Physical Strategies for Rate Enhancement
Intramolecular Catalysis
Consider the hydrolysis of phenylacetate. This reaction, a nucleophilic substitution reaction, could be catalyzed by adding the general base acetate to the solution, as described above. Since the reaction rate doubles with a doubling of the solution acetate concentration, the reaction is bimolecular (first order in reactant and catalyst). Now consider the same reaction only when the general base part of the catalyst, the carboxyl group, is part of the reactant phenylacetate. Such a case occurs in the acetylated form of salicylic acid—i.e., aspirin. When the carboxy group is ortho to the acetylated phenolic OH, it is in the perfect position to accept a proton from water, thereby decreasing the charge development on the O atom in the transition state. The general base does not have to diffuse to the appropriate site when it is intramolecular to the carbonyl C of the ester linkage. The rate of this intramolecular base catalysis is about 100-fold greater than that of an intermolecular base catalyst like acetate. It is as if the effective concentration of the intramolecular carboxyl base catalyst is much higher due to its proximity to the reaction site.
Another type of reaction involving a carboxyl group (in addition to simple proton transfer) is when the negatively charged carboxyl O acts as a nucleophile and attacks an electrophilic carbonyl carbon. When the carbonyl is part of an ester, the carboxyl group engages in a nucleophilic substitution reaction, expelling the alcohol part of the ester as a leaving group. The remaining examples below consider the nucleophilic (carboxyl) substitution on phenylesters, with phenolate as the leaving group. The reactions, in effect, transfer an acyl group to the carboxyl group, forming an anhydride.
First, consider acyl transfer with aspirin derivatives. As you know, aspirin contains a carboxyl group ortho to an ester substituent. Hence, the carboxyl group can act as a nucleophile and attack the carbonyl carbon of the ester in a nucleophilic substitution reaction. The net effect is to transfer the acetyl group from the phenolic OH to the carboxyl group, forming an anhydride. This is an intramolecular reaction. Compare this reaction to a similar bimolecular reaction shown in Figure \(\PageIndex{21}\).

The first-order rate constant of the intramolecular transfer of the acetyl group to the carboxyl group is k1 = 0.02 s-1. The analogous bimolecular reaction rate constant k2 ~ 10-10 M-1s-1. Dividing k1/k2 gives the relative rate enhancement of the intramolecular over the intermolecular reaction. With units of molarity, this ratio can be interpreted as the relative effective concentration of the intramolecular nucleophile. This makes the effective concentration of the carboxylate in the aspirin derivative 2 x 107 M.
Now consider the cleavage of phenylacetate using acetate as the nucleophile, as shown in Figure \(\PageIndex{22}\). The products are acetic anhydride and phenolate. This is a bimolecular reaction (a slow one at that), with a bimolecular rate constant k2, which is set to 1 for comparison with similar reactions.

Now consider a monoester derivative of succinic acid - phenyl succinate - in which the free carboxyl group of the ester attacks the carbonyl carbon of the ester derivative, as shown in Figure \(\PageIndex{23}\).

Suppose you assign a second-order rate constant k2 = 1 M-1s-1 to the analogous intermolecular reaction of acetate with phenylacetate (as described above). Then, the first-order rate constant for the intramolecular reaction of phenylsuccinate is 105 s-1. The ratio of rate constants, k1/k2 = 105 M. That is, it would take a 105 M concentration of acetate to react with 1 M phenylacetate in the first bimolecular reaction to match the rate of the intramolecular reaction of phenylsuccinate. The intramolecular reaction of an even more sterically restricted bicyclic phenylcarboxylate shown in Figure \(\PageIndex{24}\) has a k1/k2 = 108 M.

Another example is anhydride formation between two carboxyl groups. The ΔGo for this reaction is positive, suggesting an unfavorable reaction. Consider two acetic acid molecules condensing to form acetic anhydride. For this intermolecular reaction, Keq = 3x10-12 M-1. Now consider the analogous intramolecular reaction of the dicarboxylic acid succinic acid. It condenses in an intramolecular reaction to form succinic anhydride with a Keq = 8x10-7 (no units). The ratio Keq-intra/Keq inter = 3 x105 M. It is as if the effective concentration of the reacting groups, because they do not have to diffuse together to react, is 3 x105 M.
How does this apply to enzyme-catalyzed reactions? Enzymes bind substrates in physical steps, which are typically fast. The slow step is often the chemical conversion of the bound substrate, which is effectively intramolecular if the initial binding reaction is fast. These three kinds of reactions, intermolecular, intramolecular, and enzyme-catalyzed, can be broken down into two hypothetical steps: a binding followed by catalysis, as shown in Figure \(\PageIndex{25}\).

Suppose the rate constants for the chemical steps are all identical. Then, the advantages of the intramolecular and enzyme-catalyzed reaction over the intermolecular reaction are KINTRA/KINTER and KENZ/KINTER, respectively.
The advantage of intramolecular reactions is evident in the Ca-EDTA complex. Calcium in solution exists as an octahedral complex, with water occupying all coordination sites. EDTA, a multidentate ligand, first interacts through one of its potential six electron donors to Ca in a reaction entropically disfavored from the Ca-EDTA perspective, although one water molecule is released. Once this first intramolecular complex is formed, the rest of the ligands on the EDTA rapidly coordinate with the Ca and release bound water, as illustrated in Figure \(\PageIndex{26}\). The former is no longer entropically disfavored, as it is now an intramolecular process, whereas the latter is favored by releasing the remaining five water molecules.

Figure \(\PageIndex{26}\): Binding of Ca2+ and EDTA
We've shown above the catalytic advantage of intramolecular reactions, in terms of a dramatic increase in the effective concentration of reactants, which can reach 108 M. Another way to look at it is to consider the entropy changes associated with dimer formation. The table below shows that an intramolecular reaction is favored over an intermolecular reaction since, in the latter, significant decreases in translational and rotational entropy result.
| System | A | B | A-B | ΔS |
|---|---|---|---|---|
| Gas | ||||
| S trans | 30 | 30 | 30 | -30 |
| S rot | 20 | 20 | 20 | -20 |
| S int | 5 | 5 | 20 | +10 |
| Gas → Solution | -10 | -10 | -15 | |
| S sol | 45 | 46 | 55 | -35 (Corresponds to 108-109 M) |
Strain Distortion
In organic chemistry, you learned that certain structures, such as three-membered and four-membered ring structures, such as epoxides, were highly reactive due to the strain distortion inherent to the unfavored bond angles of the ring. Enzyme active sites can also utilize strain distortion within a bound substrate to increase the molecule's reactivity and favor the formation of the transition state. Many enzymes that function by the induced-fit model also utilize strain distortion in their catalytic mechanism. In the unbound state, they remain in a low catalytic state; however, interaction with the substrate destabilizes the enzyme's active site or induces strain within the substrate, initiating catalytic activity.
New ideas have emerged that quantitatively partition the overall catalytic enhancement conferred by enzymes into discrete features and account for the full rate enhancement. We will discuss this in Chapter 6.5, using a class of enzymes called serine proteases as an example.
A Note on Asymmetric Catalysis/Organocatalysis
In a subsequent section, we will discuss how protein enzymes use the catalytic strategies described above. An intriguing question arises: how much of the structure of a large protein is really needed for catalysis? Much work has been directed to developing small-molecule catalysis mimetics of large protein enzymes. How small can you reduce the size of a protein and still get catalysis?
One important feature of enzyme catalysis is that it catalyzes reactions that produce only one enantiomer. That is, the synthesis is asymmetric. This is typically a consequence of the asymmetric enzyme (itself chiral) binding only one enantiomer as a reactant and/or the imposition of steric restrictions on the possible reactions of the bound substrate. L-Pro alone can act as an asymmetric catalyst in an aldol condensation reaction. Figure \(\PageIndex{27}\):

Catalysts are vital in biological settings and the laboratory synthesis of molecules that sustain our culture and economy. Transition metal and, increasingly, protein enzymes have been used as industrial catalysts. New asymmetric catalysts have now joined them (a subset of organocatalysts). The work of Benjamin List and David MacMillan, who were instrumental in developing the ideas of asymmetric catalysts, has been recognized by the Nobel Commission, which awarded them the 2021 Nobel Prize in Chemistry.
The enzyme triose phosphate isomerase catalyzes an asymmetric reaction in which only one enantiomer of glyceraldehyde-3-phosphate is produced from the achiral dihydroxyacetone phosphate. Figure \(\PageIndex{28}\) shows this enantiospecific reaction.

Figure \(\PageIndex{28}\): Reaction catalyzed by triose phosphate isomerase
Figure \(\PageIndex{29}\)s contains an interactive iCn3D model of a triose phosphate isomerase from Trypanosoma brucei brucei (1KV5), which shows a conserved active site Pro 168 (spacefill) and amino acid side chains within 4 Å (stick) within the context of one monomer (cartoon) of the dimeric protein.
AsymCat.png?revision=1&size=bestfit&width=266&height=212)
Summary
(Summary written by Claude, Sonnet 4.6, Anthropic)
This chapter establishes the chemical and physical principles underlying all catalysis — enzymatic and otherwise — by examining how specific molecular strategies lower activation energy, stabilize transition states, and accelerate reactions that would otherwise be prohibitively slow under physiological conditions. The central insight is that enzymes achieve rate enhancements of 10⁷ or more not through any single mechanism but through the synergistic deployment of multiple chemical and physical strategies within a precisely organized active site.
The kinetic and thermodynamic problem is introduced through ester hydrolysis: the resonance stabilization of the ester makes the carbonyl carbon only weakly electrophilic, and water is a weak nucleophile. Hence, the uncatalyzed reaction is extremely slow. In the transition state, charge develops and separates — the carbonyl O becomes partially negative, and the attacking O becomes partially positive — creating an unstable, high-energy species. Any strategy that stabilizes the charges developing in this transition state lowers its energy and accelerates the reaction. This principle applies universally: the Hammond postulate tells us that the transition state for an endergonic step resembles the intermediate (which carries full charges), so stabilizing the intermediate also stabilizes the transition state.
General acid/base catalysis is the most common chemical strategy in enzymes. In specific acid/base catalysis, only H₃O⁺ or OH⁻ is effective, and the reaction rate is independent of buffer composition. In general acid catalysis, any proton donor (buffer acid) can donate a proton to the developing negative charge in the transition state, reducing charge separation and lowering activation energy; in general base catalysis, any proton acceptor removes the proton from the attacking nucleophile, increasing its nucleophilicity. Enzymes exploit the pKa values of amino acid side chains — Asp and Glu as general acids (~pKa 4), His imidazole as a general acid or base near pH 7, Lys as a general acid (~pKa 10) — to achieve general acid/base catalysis that is precisely tuned to the microenvironment of the active site.
Metal ion (electrostatic) catalysis exploits the Lewis acid character of transition metals (Zn²⁺, Cu²⁺, Mg²⁺, Fe²⁺/³⁺) to stabilize developing negative charges in transition states and intermediates. In the decarboxylation of β-keto acids, Cu²⁺ coordinates to the β-keto carbonyl, increasing its electrophilicity and making it a better electron sink for the departing carboxylate. In carbonic anhydrase — among the fastest enzymes known (kcat = 10⁶ s⁻¹) — Zn²⁺ coordinates three histidine side chains and a water molecule, reducing the water pKa from 14 to ~9 and generating a bound OH⁻ that attacks CO₂ at physiological pH. Given that ~1/3 of all enzymes require metal cofactors, metal ion catalysis is one of the most biologically widespread mechanisms.
Covalent (nucleophilic) catalysis changes the reaction pathway itself by introducing a new covalent intermediate. A nucleophilic catalyst — better than the ultimate nucleophile — attacks the substrate to form a transient covalent adduct, splitting the original high-energy single transition state into two lower ones, each with a lower activation energy. Pyridine catalyzes anhydride hydrolysis this way; enzymes use active site nucleophiles (Cys, Ser, His, Lys) analogously. Iminium ions (Schiff bases) formed by reaction of an active site lysine ε-amino group with a ketone or aldehyde substrate are a recurring biochemical motif: the iminium nitrogen carries a full positive charge (pKa ~7, so protonated and deprotonated forms coexist at physiological pH) and acts as a superior electron sink compared to a carbonyl, facilitating decarboxylation of β-keto acids and C–C bond cleavage reactions. Electron flow can be visualized as moving from a source (the carboxyl group) to a sink (the iminium N), a conceptual framework that simplifies mechanistic analysis. Aldolase provides a textbook example where Lys239 forms an actual Schiff base with fructose-1,6-bisphosphate in the crystal structure.
Transition state stabilization is Pauling's elegant thermodynamic formulation of all catalysis: an enzyme need only bind the transition state more tightly than the substrate to lower the activation energy. Using a linked equilibrium diagram and the thermodynamic principle that ΔG° is a state function, one derives KT/KS = kE/kN, where KT and KS are the equilibrium constants for enzyme binding of S‡ and S, respectively, and kE and kN are the catalyzed and uncatalyzed first-order rate constants. Since kE/kN ~ 10⁸–10¹⁴, and KS ~ 10³–10⁵ M⁻¹ for typical substrates, KT ~ 10¹⁵ M⁻¹ — a dissociation constant of ~10⁻¹⁵ M (femtomolar), comparable to the avidin-biotin interaction. This framework explains why stable transition state analogs (molecules that mimic the geometry and charge distribution of the transition state without undergoing the reaction) are among the most potent enzyme inhibitors known — and why they are powerful starting points for drug design. Abzymes (catalytic antibodies) provide direct experimental proof: immunizing mice with a phosphonate TS analog of an ester (sp³ phosphorus mimicking the sp³ carbon of the tetrahedral intermediate, with an oxyanion-like negative oxygen) elicits antibodies that catalyze ester hydrolysis, demonstrating that any binding site that recognizes the transition state geometry will catalyze the reaction.
Physical strategies complement chemical ones. Intramolecular catalysis eliminates the entropic cost of bimolecular reactions — the loss of translational and rotational entropy (~35 cal/K·mol in solution) when two molecules must diffuse together and orient correctly. The ratio of intramolecular to intermolecular rate constants (k₁/k₂, in units of M) quantifies the effective concentration advantage: ~100-fold for aspirin's ortho-carboxyl acting as a general base, ~10⁵ M for phenyl succinate, and ~10⁸ M for bicyclic phenylcarboxylate systems. Enzyme-bound substrates experience this same advantage because the chemical step after binding is effectively intramolecular; the entropic cost was paid during the fast binding step. Strain distortion in induced-fit enzymes destabilizes the ground state of the enzyme-substrate complex toward the transition state geometry, further lowering the activation barrier. Finally, asymmetric (organo)catalysis illustrates that much of what enzymes do can, in principle, be replicated by small molecules: L-proline alone catalyzes asymmetric aldol condensations through an iminium ion mechanism, producing a single enantiomeric product — the same strategy used by aldolase. The development of asymmetric small-molecule catalysts, recognized by the 2021 Nobel Prize in Chemistry, represents a convergence of chemical and biochemical principles with practical applications in pharmaceutical synthesis.
References
Amyes TL, Richard JP. Biochemistry. 2013, 52(12), 2021-35. doi: 10.1021/bi301491r
Ferst and Kirby, J. Am. Chem. Soc. 1967, 89, 19, 4857–4863. https://doi-org.ezproxy.csbsju.edu/10.1021/ja00995a007
Komiyama et al., PNAS, 1977, 74 (7) 2634-2638. https://doi.org/10.1073/pnas.74.7.263
Lerner, R. A., & Tramontano, A. (1987). Antibodies as enzymes. Trends in Biochemical Sciences, 12, 427-430. https://doi.org/10.1016/0968-0004(87)90208-8
OpenStax College. (2022). Chemistry. OpenStax. https://openstax.org/details/books/chemistry-2e
Regan, C. K., Craig, S. L., & Brauman, J. I. Science. 2002 295(5563), 2245-2247. DOI: 10.1126/science.1068849