
8.1: Nucleic Acids - Structure and Function
- Page ID
- 14963


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Differentiate Between DNA and RNA Structures:
- Explain the basic composition of nucleic acids (sugar–phosphate backbone and nitrogenous bases).
- Describe how the sugar (deoxyribose in DNA vs. ribose in RNA) and the specific bases (T in DNA vs. U in RNA) influence overall structure and function.
-
Describe Canonical and Noncanonical Base Pairing:
- Illustrate the principles of Watson–Crick base pairing and how hydrogen bonding between complementary bases stabilizes the double helix.
- Define and compare alternative (noncanonical) base pairing modes, including reverse Watson–Crick, wobble, Hoogsteen, and reverse Hoogsteen base pairs, and explain how these variations affect nucleic acid function.
-
Understand DNA Conformations and Structural Dynamics:
- Compare the three main double-helical forms of DNA (A-DNA, B-DNA, and Z-DNA), focusing on differences in handedness, base pair spacing, twist angles, and groove dimensions.
- Analyze how environmental factors (e.g., hydration, salt concentration) and molecular interactions induce transitions between these forms.
-
Explore Higher-Order Nucleic Acid Structures:
- Describe the formation and biological significance of G-quadruplexes, including how guanine tetrads and metal ions (e.g., K⁺) stabilize these structures.
- Explain the mechanisms of triple helix formation, both intermolecular and intramolecular (H-DNA), and discuss their potential roles in gene regulation.
- Recognize the characteristics of four-way junctions and their role in nucleic acid folding and interactions with small molecules.
-
Analyze the Role of Base Stacking in Nucleic Acid Stability:
- Explain how base stacking and hydrophobic interactions contribute to the stability of the DNA double helix, in addition to hydrogen bonding.
- Evaluate the effects of nucleotide sequence (e.g., GC vs. AT content) on the melting temperature (Tm) and overall stability of nucleic acids.
-
Interpret Structural Models and Parameters:
- Utilize interactive molecular visualization tools (e.g., iCn3D) to explore nucleic acid structures.
- Identify key structural parameters such as propeller and twist angles and understand how they influence nucleic acid conformation and dynamics.
-
Relate Nucleic Acid Structure to Biological Function:
- Connect the structural features of DNA (e.g., major and minor grooves) with the mechanisms of protein recognition and binding.
- Discuss how conformational flexibility (e.g., local “breathing” and dynamic unfolding) enables critical cellular processes like replication, transcription, and repair.
-
Evaluate Experimental Approaches in Nucleic Acid Research:
- Summarize the historical and modern techniques used to elucidate nucleic acid structure, including X-ray crystallography, NMR, and cryo-electron microscopy.
- Analyze how advances in structural biology have deepened our understanding of nucleic acid dynamics and their roles in cellular function.
These goals will help you integrate knowledge of nucleic acid chemistry with its biological implications, preparing you for advanced study and research in biochemistry.
Introduction to Nucleic Acids
Alongside proteins, lipids, and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life. The nucleic acids consist of two major macromolecules, Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) that carry the genetic instructions for the development, functioning, growth, and reproduction of all known organisms and viruses. Both consist of polymers of a sugar-phosphate-sugar backbone with organic heterocyclic bases attached to the sugars. The sugar in DNA is deoxyribose while in RNA it is ribose. DNA contains four bases, cytosine and thymine (pyrimidine bases) and guanine and adenine (purine bases). DNA in vivo consists of two antiparallel strands intertwined to form the iconic DNA double-stranded helix. RNA is single-stranded but may adopt many secondary and tertiary conformations not unlike that of a protein. Figure \(\PageIndex{1}\) shows a low-resolution comparison of the structure of DNA and RNA.

The biological function of DNA is quite simple: to carry and protect the genetic code. Its structure serves that purpose well. In the next section, we will study the functions of RNA, which are much more numerous and complicated. The structure of RNA has evolved to serve those added functions.
The core structure of a nucleic acid monomer is the nucleoside, which consists of a sugar residue + a nitrogenous base that is attached to the sugar residue at the 1′ position as shown in Figure \(\PageIndex{2}\). The sugar utilized for RNA monomers is ribose, whereas DNA monomers utilize deoxyribose, which has lost the hydroxyl functional group at the 2′ position of ribose. DNA contains four nitrogenous bases, including the Purines, Adenine (A), and Guanine (G), and the Pyrimidines, Cytosine (C), and Thymine (T). RNA uses the same nitrogenous bases as DNA, except for Thymine. Thymine is replaced with Uracil (U) in the RNA structure.
When one or more phosphate groups are attached to a nucleoside at the 5′ position of the sugar residue, it is called a nucleotide. Nucleotides come in three flavors depending on how many phosphates are included: the incorporation of one phosphate forms a nucleoside monophosphate, the incorporation of two phosphates forms a nucleoside diphosphate, and the incorporation of three phosphates forms a nucleoside triphosphate as shown in Figure \(\PageIndex{2}\).

DNA and RNA Hydrogen-bonded structures
Figure \(\PageIndex{3}\) below shows a "flattened" structure of double-stranded B-DNA that best shows the backbone and hydrogen-bonded base pairs between two antiparallel strands of the DNA. Unlike the protein α-helix, where the R-groups of the amino acids are positioned to the outside of the helix, in the DNA double-stranded helix, the nitrogenous bases are positioned inward and face each other. The backbone of the DNA is made up of repeating sugar-phosphate-sugar-phosphate residues. Bases fit in the double-helical model if pyrimidine on one strand is always paired with purine on the other. From Chargaff’s rules, the two strands will pair A with T and G with C. This pairs a keto base with an amino base and a purine with a pyrimidine. Two H‑bonds can form between A and T, and three can form between G and C. This third H-bond in the G:C base pair is between the additional exocyclic amino group on G and the C2 keto group on C. The pyrimidine C2 keto group is not involved in hydrogen bonding in the A:T base pair.
Furthermore, the orientation of the sugar molecule within the strand determines the directionality of the strands. The phosphate group that makes up part of the nucleotide monomer is always attached to the 5′ position of the deoxyribose sugar residue. The free end that can accept a new incoming nucleotide is the 3′ hydroxyl position of the deoxyribose sugar. Thus, DNA is directional and is always synthesized in the 5′ to 3′ direction. Interestingly, the two strands of the DNA double helix lie in opposite directions or have a head-to-tail orientation.

By analogy to proteins, DNA and RNA can be loosely thought to have primary and secondary structures. For a single strand, the primary sequence is just the base sequence read from the 5' to 3' end of the strand, with the bases thought of as "side chains" as illustrated in Figure \(\PageIndex{4}\) for an RNA strand which contains U instead of T.
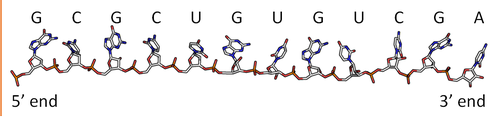
Since it is found partnered with another molecule (strand) of DNA, the double-stranded DNA, which consists of two molecules held together by hydrogen bonds, might be considered to have secondary structure (analogous to alpha and beta structure in proteins). Of course, the hydrogen bonds are not between backbone atoms but between side chain bases in double-stranded DNA.
Figure \(\PageIndex{5}\) shows an interactive iCn3D model of the iconic structure of a short oligomer of double-stranded DNA (1BNA).
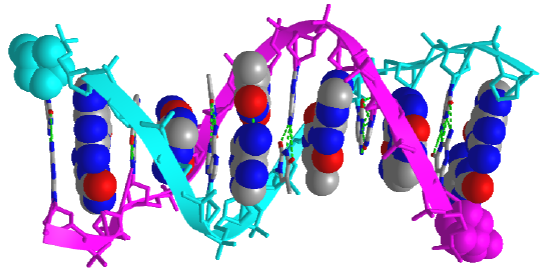
The backbones of the antiparallel strands are magenta (chain A) and cyan (chain B). Each chain's 5' sugar-phosphate end is shown in spacefill and colored magenta (chain A) and cyan (chain B). The hydrogen-bonded interstrand base pairs are shown in two ways: as spacefill and sticks to illustrate how the bases stack over each other.
Figure \(\PageIndex{6}\) shows types of "secondary (flat representations) and their 3D or tertiary representations found in nucleic acids.
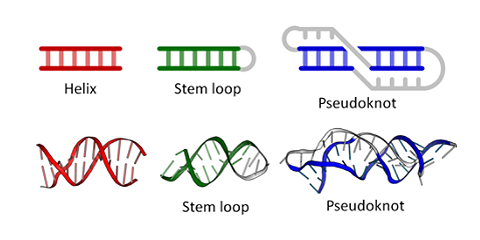
Figure \(\PageIndex{7}\) shows an interactive iCn3D model of the tertiary structure of the T4 hairpin loop on a Z-DNA stem (1D16).
.png?revision=1&size=bestfit&width=387&height=272)
The hairpin shown is from a synthetic DNA oligomer C-G-C-G-C-G-T-T-T-T-C-G-C-G-C-G, which adopts an alternative Z-DNA conformation (which we will explore below) with a loop at one end. The thymine bases 7, 8, and 9 are generally perpendicular to one another and stack together, along with the ribose of T7.
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of pseudoknot in RNA (437D).
.png?revision=1&size=bestfit&width=335&height=259)
The pseudoknot has two stems that form a "helix" and two loops. The knot consists of a hairpin in the nucleic acid structure, with the loop between the helices paired to another part of the nucleic acid. Pseudoknots can be found in mRNA and ribosomal RNA and affect the translation of the RNA (decoding to instruct the synthesis of a protein sequence). RNA viruses have pseudoknots, which likewise affect protein synthesis and RNA replication. Pseudoknots also occur in DNA.
Synthesis and structure of DNA
The nucleotide that is required as the monomer for the synthesis of both DNA and RNA is nucleoside triphosphate. During the incorporation of the nucleotide into the polymeric structure, two phosphate groups (PPi, called pyrophosphate) from each triphosphate are cleaved from the incoming nucleotide and further hydrolyzed during the reaction, leaving a nucleoside monophosphate that is incorporated into the growing RNA or DNA chain as shown in Figure \(\PageIndex{9}\) below. The nucleophilic attack of the 3′-OH of the growing DNA polymer mediates the attack on the incoming nucleoside triphosphate. Thus, DNA synthesis is directional, only occurring at the 3′-end of the molecule.
The further hydrolysis of the pyrophosphate (Pi-Pi) releases a large amount of energy, ensuring that the overall reaction has a negative ΔG. Hydrolysis of Pi-Pi ↔ 2Pi has a ΔG = -7 kcal/mol (-29 kJ/mol) and is essential to provide the overall negative ΔG (-6.5 kcal/mol, -27 kJ/mol) of the DNA synthesis reaction. Hydrolysis of the pyrophosphate also ensures that the reverse reaction, pyrophosphorolysis, will not occur, removing the newly incorporated nucleotide from the growing DNA chain.
This reaction is mediated in DNA by a family of enzymes known as DNA polymerases. Similarly, RNA polymerases are required for RNA synthesis. A more detailed description of polymerase reaction mechanisms will be covered in Chapter 24, which deals with DNA replication and repair, and DNA transcription in Chapter 25.

DNA was first isolated by Friedrich Miescher in 1869. The double-helix model of DNA structure was first published in the journal Nature by James Watson and Francis Crick in 1953 based upon the crucial X-ray diffraction image of DNA from Rosalind Franklin in 1952, followed by her more clarified DNA image with Raymond Gosling, Maurice Wilkins, Alexander Stokes, and Herbert Wilson, and base-pairing chemical and biochemical information by Erwin Chargaff. The prior model was triple-stranded DNA.
The realization that the structure of DNA is that of a double helix elucidated the mechanism of base pairing by which genetic information is stored and copied in living organisms and is widely considered one of the most important scientific discoveries of the 20th century. Crick, Wilkins, and Watson each received one-third of the 1962 Nobel Prize in Physiology or Medicine for their contributions to the discovery. (Franklin, whose breakthrough X-ray diffraction data was used to formulate the DNA structure, died in 1958 and thus was ineligible to be nominated for a Nobel Prize.)
Watson and Crick proposed two strands of DNA – each in a right-hand helix – wound around the same axis. The two strands are held together by H-bonding between the complementary base pairs (A pairs with T and G pairs with C) as shown in Figure \(\PageIndex{10}\) below. Note that when looking from the top view, down on a DNA base pair, the position where the base pairs attach to the DNA backbone is not equidistant, but that attachment favors one side over the other. This creates unequal gaps or spaces in the DNA known as the major groove for the larger gap and the minor groove for the smaller gap (Figure 4.5). Based on the DNA sequence within the region, the hydrogen-bond potential created by the nitrogen and oxygen atoms present in the nitrogenous base pairs causes unique recognition features within the major and minor grooves, allowing for specific protein recognition sites to be created.

Figure \(\PageIndex{1}\) shows a schematic representation of available hydrogen bond donors and acceptors in the major and minor grooves for TA and CG base pairs.

Figure \(\PageIndex{12}\) shows an interactive iCn3D model of DNA showing the major and minor grooves.
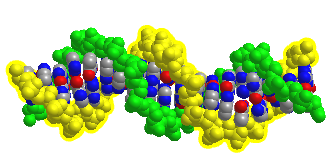
The two sugar-phosphate backbones are shown in green and yellow. Some of the red (oxygen) and blue (nitrogen) atoms in the major groove (and to a much lesser extent in the minor groove) are not involved in inter-strand G-C and A-T base pairing and so would be available to hydrogen bond donors with specific binding proteins that would display complementary shape and hydrogen bond acceptors and donors. (The white spheres are Cd ions.)
Figure \(\PageIndex{13}\) shows an interactive iCn3D model of the N-terminal fragment of the yeast transcriptional activator GAL4 bound to DNA (1D66).
.png?revision=1&size=bestfit&width=449&height=250)
The N-terminal fragment binds to conserved CCG triplets found at both ends of the DNA in the major groove. The protein shown is a dimer held together by a short coiled-coil interaction domain, so the site has 2-fold symmetry. A small Zn2+-containing secondary structure motif in each member of the dimer interacts with the major groove. An extended chain connects the DNA-binding and interaction domains of each protein.
In addition to the major and minor grooves providing variation within the double helix structure, the axis alignment of the helix, along with other influencing factors such as the degree of solvation, can give rise to three forms of the double helix, the A-form (A-DNA), the B-form (B-DNA), and the Z-form (Z-DNA) as shown in Figure \(\PageIndex{14}\).

Both the A- and B-forms of the double helix are right-handed spirals, with the B-form being the predominant form found in vivo. The A-form helix arises when conditions of dehydration below 75% of normal occur and has mainly been observed in vitro during X-ray crystallography experiments when the DNA helix has become desiccated. However, the A-form of the double helix can occur in vivo when RNA adopts a double-stranded conformation or when RNA-DNA complexes form. The 2′-OH group of the ribose sugar backbone in the RNA molecule prevents the RNA-DNA hybrid from adopting the B-conformation due to steric hindrance.
The third double helix type formed is a left-handed helical structure known as the Z-form or Z-DNA. Within this structural motif, the phosphates within the backbone appear to zigzag, providing the name Z-DNA. In vitro, the Z-form of DNA is adopted in short sequences that alternate pyrimidine and purines when high salinity is present. However, the Z-form has been identified in vivo within short regions of the DNA, showing that DNA is quite flexible and can adopt a variety of conformations. A comparison of features between A-, B-, and Z-form DNA is shown in Table 4.1.
| B-DNA | A-DNA | Z-DNA | |
|---|---|---|---|
| helix sense | Right Handed | Right Handed | Left Handed |
| base pairs per turn | 10 | 11 | 12 |
| vertical rise per bp | 3.4 Å | 2.56 Å | 19 Å |
| rotation per bp | +36° | +33° | -30° |
| helical diameter | 19 Å | 19 Å | 19 Å |
The double-stranded helix of DNA is not always stable. This is because the attractions between the strands are noncovalent, reversible interactions. Depending on the DNA sequence, denaturation (melting) can be local or widespread, enabling various crucial cellular processes, including DNA replication, transcription, and repair.
Both sequence specificity and interaction (whether covalent or not) with a small compound or a protein can induce tilt, roll, and twist effects that rotate the base pairs in the x, y, or z axis, respectively, as seen in Figure \(\PageIndex{15}\), and can therefore change the helix’s overall organization. Furthermore, slide or flip effects can also modify the geometrical orientation of the helix. Hence, the flip effects and (to a lesser extent) the other movements described above modulate the double-strand stability within the helix or at its ends. Indeed, under physiological conditions, local DNA ‘breathing’ has been evidenced at both ends of the DNA helix, and B-DNA to Z-DNA structural transitions have been observed in internal DNA regions. These locally open DNA structures are good substrates for specific proteins, which can also induce the opening of a ‘closed’ helix. The DNA replication and repair processes will be discussed in more detail in Chapter 24.

Figure \(\PageIndex{15}\): Localized Structural Modification of the DNA Double Helix. (a) Base pair orientations with the x, y, and z axes result in different kinds of rotation (tilt, roll, or twist) or slipping of the bases (slide, flip) regarding the central helix axis. (b) Mature B-DNA has nearly 11 base pairs within one helical turn. (c) Mono- or bis-intercalation of a small molecule (shown in blue) between adjacent base pairs, resulting in an unwinding of the DNA helix (orange arrow on the top) and a lengthening of the DNA helix (ΔLength) depending on the X and y Å values that are specific for a defined DNA intercalating compound. (d) Representation of the DNA bending, base flipping, or double-strand opening induced by some DNA destabilizing alkylating agents (adducts shown in blue). Adapted from Calladine and Drew’s schematic box representation. Lenglet and David-Cordonnier (2010) Journal of Nucleic Acids, http://dx.doi.org/10.4061/2010/290935. Creative Commons Attribution License,
Figure \(\PageIndex{16}\) shows interactive iCn3D models of A-DNA (top), B-DNA (center), and Z-DNA (bottom). (Copyright; author via source). Click the image for a popup or use the external links in column 1.
| A-DNA (440D) | 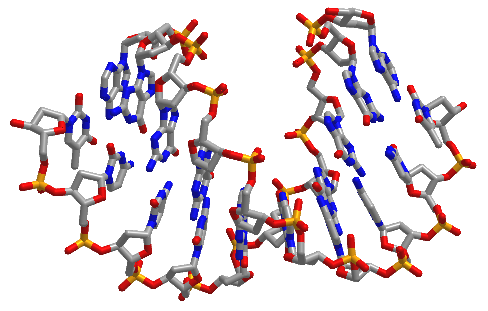 |
| B-DNA (1BNA) | 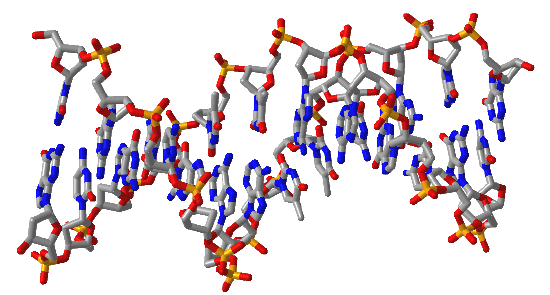 |
| Z-DNA (4OCB) | 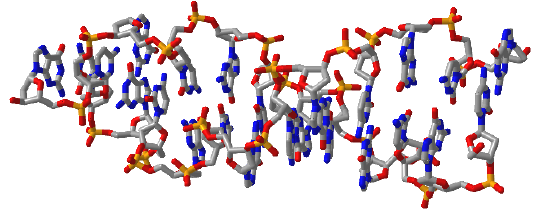 |
 Figure \(\PageIndex{16}\): A, B, and Z-DNA. Click the image for a popup or use the links in column 1
Figure \(\PageIndex{16}\): A, B, and Z-DNA. Click the image for a popup or use the links in column 1
We studied the structure of proteins in-depth, discussing resonance in the peptide backbone, allowed backbone angles φ, ψ, and ω, side chain rotamers, Ramachandran plots, and different structural motifs. We also explored them dynamically using molecular dynamics simulations. We also discussed the thermodynamics of protein stability and how stability could be altered by changing environmental factors such as solution composition and temperature.
In contrast, our understanding of the structural parameters and the dynamics of nucleic acids is less advanced. This may seem paradoxical, especially given the apparent simplicity of the iconic structure of DNA presented in textbooks. Yet, we should look at the types of secondary structures of nucleic acids presented and then the complicated tertiary and quaternary structures of RNA.
The nucleic acid backbone has a 5-membered sugar ring, which adds rigidity to the backbone, linked to another sugar ring by CH2O(PO3)O- connectors, adding some additional conformational freedom. We'll explore the effects of the pentose ring geometry in RNA and DNA in Chapter 8.3. To illustrate a yet unexplored complexity of nucleic acid structure, consider just the orientation of rings in double-stranded DNA and in regions of RNA where double-stranded structures form. The variants in the orientation of the hydrogen-bonded base pairs and the corresponding parameters that define them are shown in Figure \(\PageIndex{17}\).
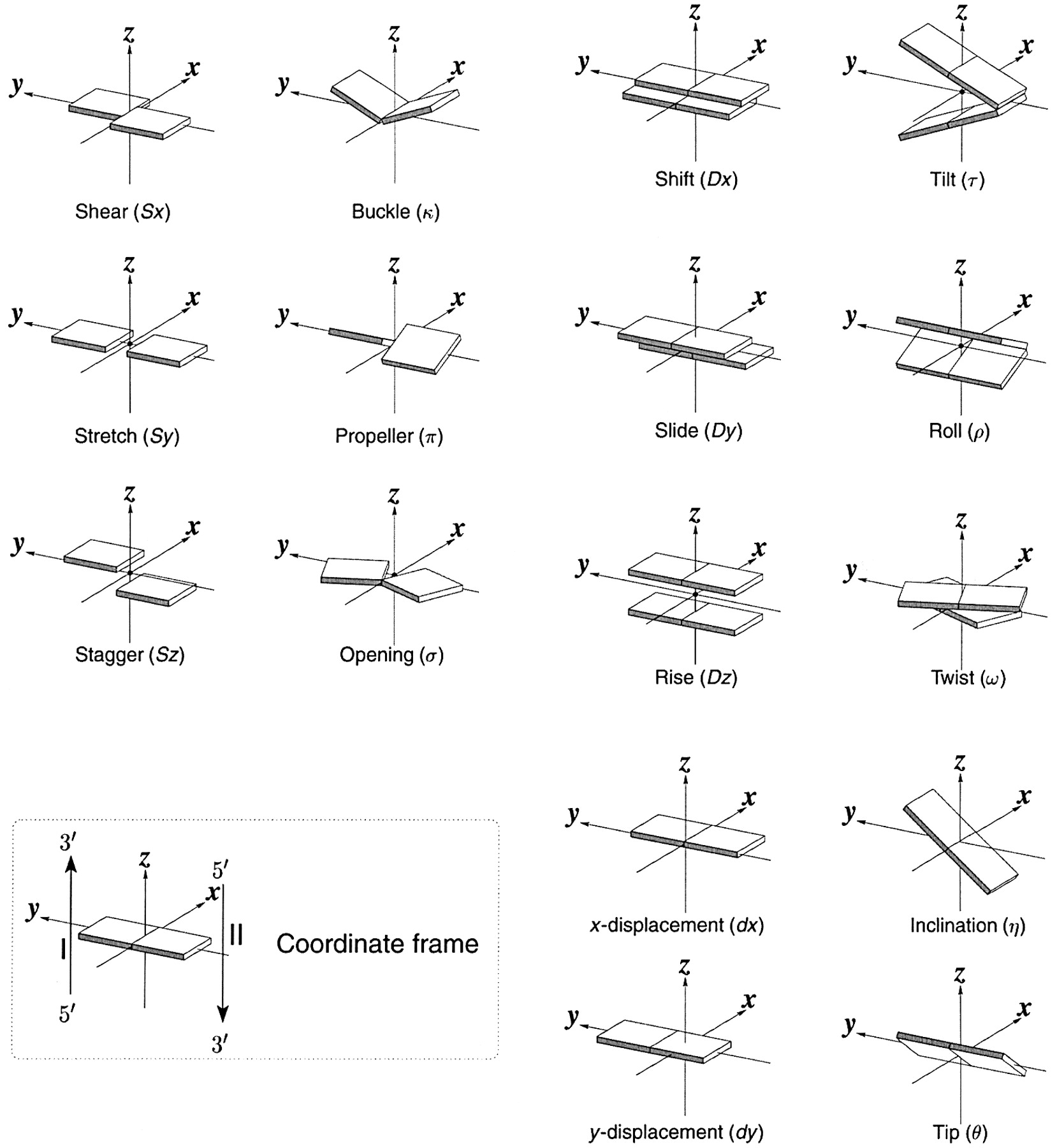
Figure \(\PageIndex{17}\): Base pair orientation and corresponding parameters in nucleic acids. http://x3dna.org/highlights/schemati...air-parameters (with permission). 2008 3DNA Nature Protocols paper (NP08), the initial 3DNA Nucleic Acids Research paper .
Consider just two of these: the propeller and twist angles. If you examine the iCn3D models of nucleic acids presented above, you will see that the base pairs are not perfectly flat but are twisted. Larger propeller angles are associated with increased rigidity. The propeller angles for A, B, and Z DNA are + 18o, + 16 +/-7 o, and about 0 o, respectively. The twist angles A, B, and Z DNA are +33 o, +36 o, and -30 o, respectively. The lower the twist angle, the higher the number of base pairs per turn. This, of course, affects the pitch of the helix (the length of one complete turn). These terms should be minimized to computationally determine the lowest energy state for a given double-stranded nucleic acid.
Alternative Base Pairing in DNA and RNA
A first glance at a DNA or RNA structure reveals a myriad of possible hydrogen bond donors and acceptors in the nucleic acid bases. Hence, it should come as no surprise that a variety of alternative or noncanonical (not in the canon or dogma) intermolecular hydrogen bonds can form between and among bases, leading to alternatives to the classical Watson-Crick base pairing. There are 28 possible base pairs with two hydrogen bonds between them. As structure determines function and activity, these alternative structures influence DNA/RNA function. We will consider four types of noncanonical base pairing: reverse Watson-Crick, wobble, Hoogsteen, and reverse Hoogsteen base pairs.
These noncanonical base pairs can occur in DNA when bases become mismatched in double-stranded regions. In RNA, which we will explore more fully in Chapter 8.2, double-stranded molecules formed by separate RNA molecules aren't common. Instead, the molecule folds on itself in 3D space to form a complex tertiary structure containing regions of helical secondary structure. RNAs also form quaternary structures when bound to other nucleic acids and proteins. Larger RNAs have loops with complex secondary and tertiary structures, which often require noncanonical base pairing, stabilizing the alternative structures. The noncanonical structures are also important for RNA-protein interactions in the RNA region that binds proteins. As with protein-ligand interactions, protein binding to RNA might also induce conformation changes, specifically noncanonical base pairs, in the RNA. For example, the HIV Rev peptide binds to a target site in the envelope gene of HIV (which has an RNA genome) and leads to the formation of an RNA loop with hydrogen bonding between two purines.
Figure \(\PageIndex{18}\) shows an interactive iCn3D model of the REV Response element RNA complexed with REV peptide (1ETF).
.png?revision=1&size=bestfit&height=430)
The peptide is shown in cyan, and its arginine side chains are shown as cyan lines. An extraordinary number of arginines form ion-ion interactions with the negatively charged phosphates in the major groove of this double-stranded A-RNA. The noncanonical base pairs are shown in CPK-colored sticks. A wobble base, U43-G77 (see below), can be seen, as well as three homopurine base pairs, G47-A73, G55-A58, and G48-G71. The solitary A68 base is shown projecting away from the RNA.
Figure \(\PageIndex{19}\) shows the Watson-Crick model and the first set of alternative non-canonical base pairs.

Figure \(\PageIndex{19}\): Some noncanonical base nucleic acid base pairs
Let's look at them in more detail.
Reverse Watson Crick: The reverse Watson-Crick AT (AU) and GC pairs can sometimes be found at the end of DNA strands and RNA. In forming the reverse base pairs, the pyrimidine can rotate 180 o along the axis shown and then rotate in the plane to align the hydrogen bond donors and acceptors, as shown in the top part of the figure. The glycosidic bond between the N in the base and the sugar (the circled R group) is now in an "antiparallel" arrangement in the reverse base pair.
Wobble Base Pairs
The bases in nucleic acids can undergo tautomerization to produce forms that can base pair noncanonically. They are termed wobble base pairs and include G-T(U) base pairs from keto-enol tautomerism and A-C base pairs from amino–imino tautomerism, as illustrated in Figure 18 above.
Figure \(\PageIndex{20}\) shows an interactive iCn3D model of the GT Wobble Base-Pairing in Z-DNA form of d(CGCGTG) (1VTT). Two such GT pairs are found in the structure.
_(1VTT).png?revision=1&size=bestfit&width=262&height=371)
The water around the wobble base pairs can form hydrogen bonds and stabilize the pair if a hydrogen bond is missing.
Figure \(\PageIndex{21}\) shows an interactive iCn3D model of dsRNA with G-U wobble base pairs (6L0Y).
.png?revision=1&size=bestfit&width=513&height=173)
The structure contains many GU wobble base pairs and two CU base pairs between two pyrimidine bases.
Inosine, a variant of the base adenine, can be found in RNA. It is formed by the deamination of adenosine by the enzyme adenosine deaminase. A nucleotide having inosine is named hypoxanthine. Hypoxanthine can form the wobble base pairs I-U, I-A, and I-C when incorporated into RNA, as illustrated in Figure \(\PageIndex{22}\).

Figure \(\PageIndex{22}\): Wobble bases pairs using hypoxanthine with the base inosine
Wobble base pair interactions are especially important in translation when a protein sequence is made from a messenger RNA template (which will be discussed in Unit III). For that decoding process, two RNA molecules, messenger RNA (mRNA) and a transfer RNA (tRNA) covalently attached to a specific amino acid like glutamic acid, must bind to each other through a 3-base pair interaction. The three bases on the mRNA are called the codon, and the three complementary bases on the tRNA are called the anticodon. The triplet base pairs are antiparallel to each other. The interaction between mRNA and tRNA is illustrated in Figure \(\PageIndex{23}\).

Figure \(\PageIndex{23}\): The wobble uridine (U34) of tRNA molecules that recognize both AA and AG-ending codons for Lys, Gln, and Glu, is modified by the addition of both a thiol (s2) and a methoxy-carbonyl-methyl (mcm5). This double modification enhances the translational efficiency of AA-ending codons. Goffena, J et al. Nat Commun 9, 889 (2018). https://doi.org/10.1038/s41467-018-03221-z. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
The third 3' base in the mRNA is less restricted and can form noncanonical, specifically, wobble base pairs, with the 5' base in the anti-codon triplet of tRNA. The term wobble arises from the subtle conformational changes used to optimize the pairing of the triplets. Wobble bases occur much more in tRNA than in other nucleic acids.
Hoogsteen base pairing
Flexibility in DNA allows rotation around the C1'-N glycosidic bond connecting the deoxyribose and base in DNA, allowing different orientations of AT and GC base pairs with each other. The normal "anti" orientation allows "Watson-Crick" (WC) base pairing between AT and GC base pairs, while the altered rotation allows "Hoogsteen" base pairs. Figure \(\PageIndex{24}\) shows the different orientations for an AT base pair.

Figure \(\PageIndex{24}\): Xu, Y., McSally, J., Andricioaei, I. et al. Modulation of Figure \(\PageIndex{xx}\)Hoogsteen dynamics on DNA recognition. Nat Commun 9, 1473 (2018). https://doi.org/10.1038/s41467-018-03516-1Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Hoogsteen base pairing is usually seen when DNA is distorted through interactions with bound proteins and drugs that intercalate between base pairs. Figure \(\PageIndex{25}\) shows an interactive iCn3D model of a Hoogsteen base pair embedded in undistorted B-DNA - MATAlpha2 homeodomain bound to DNA (1K61).
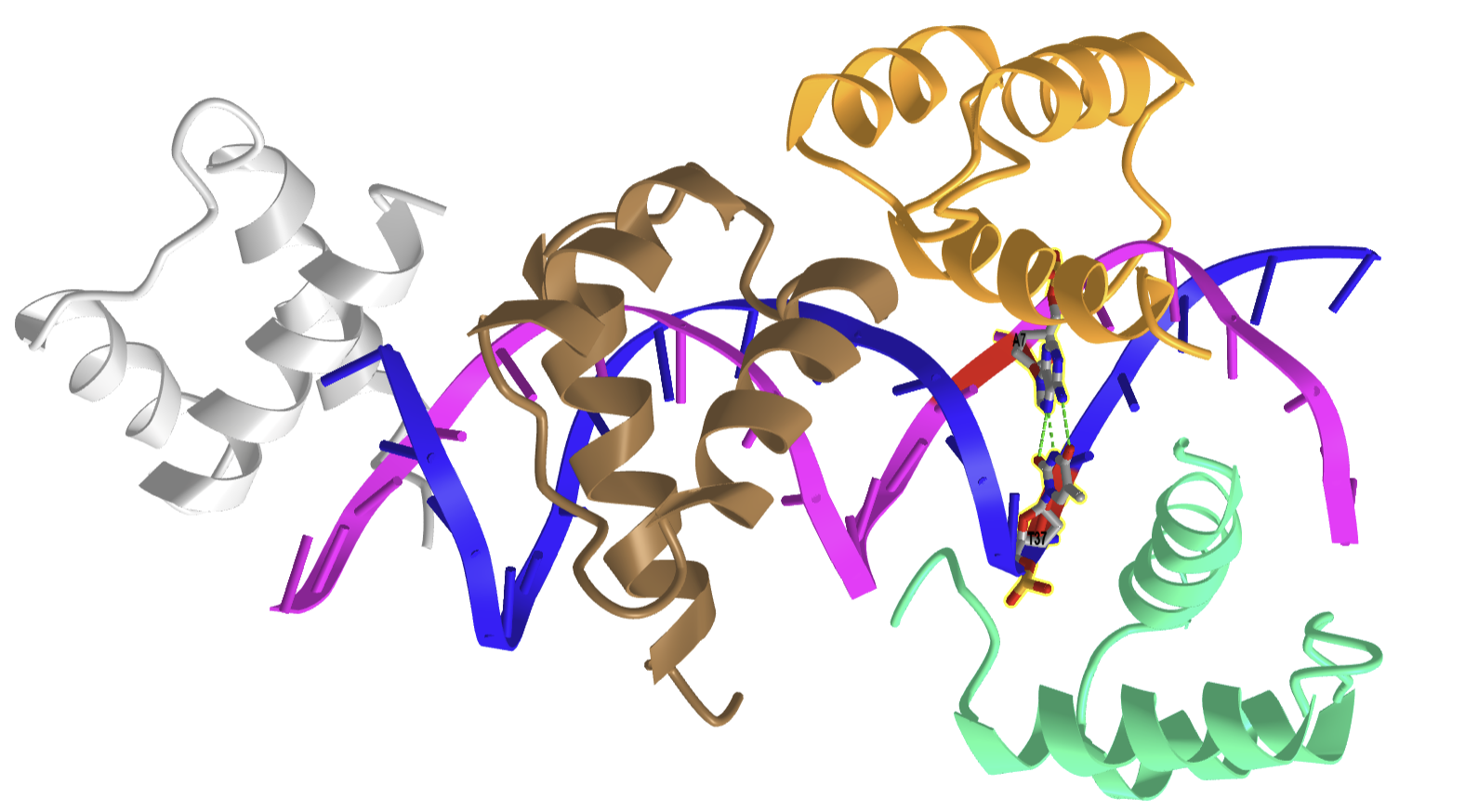.png?revision=1)
The same DNA without bound protein has no Hoogsteen base pairs. To form Hoogsteen base pairs, a rotation around the glycosidic-base bond must occur. Hoogsteen base pairs between G and C can also occur on rotation, but in addition, the N3 of cytosine is protonated, as shown in Figure 14 above.
Evidence suggests that Hoogsteen base pairing may be important in DNA replication, binding, damage, or repair. They can induce kinking of the DNA near the major groove.
There are also reverse Hoogsteen base pairing examples, as shown in Figure \(\PageIndex{26}\).

Figure \(\PageIndex{26}\): The reverse Hoogsteen AT base pair
Additional Alternative Structures: Quadruplexes, Triple Helices, and 4-Way Junctions
Quadruplexes
These can be formed in DNA and RNA from G-rich sequences involving tetrads of guanine bases that are hydrogen-bonded. They are a bit hard to describe in words, so let's first examine one particular structure.
In human cells, telomeres (the ends of chromosomes) contain 300-8000 repeats of a simple TTAGGG sequence. The repetitive TTAGGG sequences in telomeric DNA can form quadruplexes. Figure \(\PageIndex{27}\) shows an interactive iCn3D model of parallel quadruplexes from human telomeric DNA (1KF1). The structure contains a single DNA strand (5'-AGGGTTAGGGTTAGGGTTAGGG-3') which contains four TTAGGG repeats.
.png?revision=1&size=bestfit&height=315)
Figure \(\PageIndex{27}\): parallel quadruplexes from human telomeric DNA (1KF1). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...y5joFHDgWJQsQ6
Rotate the model to see three parallel layers of quadruplexes. In each layer, 4 noncontiguous guanine bases interact with a K+ ion. Hover over the guanine bases in one layer, and you will find that one layer consists of guanines 4, 10, 16, and 22, which derive from the last G in each of the repeats in the sequence of the oligomer used (5'-AGGGTTAGGGTTAGGGTTAGGG-3'). These quadruplexes certainly serve in recognition and as binding sites for telomerase proteins. The guanine-rich telomere sequences, which can form quadruplexes, may also function to stabilize chromosome ends.
A Quadruplex can be formed in 1 strand of nucleic acid (as in the above model) or from 2 or 4 separate strands. They also must have at least two stacked triads. As in the example above, single-stranded sections can form an intramolecular G-quadruplex from a GmXnGmXoGmXpGm sequence, where m is the number of Gs in each short segment (3 in the structure above). A G might be in a loop if a segment is longer than the others.
Triple Helices
These structures can occur in DNA (and also RNA) that contain homopurine and homopyrimidine sequences that have a mirror repeat symmetry. Hence, they can occur naturally. A mirror repeat contains a center of symmetry on a single strand. Here is an example: 5'-GCATGGTACG-3'.
They can also occur when a third single-stranded DNA (a triplex-forming oligonucleotide or TFO) binds to a double-stranded DNA. The TFOs bind through Hoogsteen base pairing in the major groove of the ds-DNA. They can bind tightly and specifically in a parallel or antiparallel fashion. Specific and locally higher concentrations of divalent cations or positively charged polyamines like spermine act to stabilize the extra negative charge density from the binding of a third polyanionic DNA strand.
An example of a triple helix system that has been studied in vitro is shown in Figure \(\PageIndex{28}\).

Figure \(\PageIndex{28}\): Intermolecular triplex formation and their oligonucleotide sequences (where “•” and “-” indicate Hoogsteen and Watson–Crick base pairings, respectively). Inset: chemical structure of a parallel T•AT triplet. Guerrini, L. and Alvarez-Puebla, R.A. Nanomaterials 2021, 11, 326. https://doi.org/10.3390/nano11020326. Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/)
The double-stranded canonical helix (D1D2) consists of 31 base pairs in which strand D1 is pyrimidine-rich and D2 is purine-rich strand (D2). A 22-nucleotide Triple helix-forming oligonucleotide (TFO) that is rich in pyrimidines binds the 19 AT and 2 C-GC base triplets. The TFO binds along the major groove of the D2 strand, which is purine-rich.
If the binding of the third strand in the major groove occurs at the site where RNA polymerase binds to a gene, then the third strand can inhibit gene transcription. Binding can also lead to a mutation or recombination at the site.
Figure \(\PageIndex{29}\) shows the base pairing of purine and pyrimidines of the third strand to the canonical AT dn GC base pairs of the original double-stranded DNA.

Figure \(\PageIndex{29}\): Base pairing in triple helix motifs. (after Jain et al. Biochimie. 2008. doi: 10.1016/j.biochi.2008.02.011
Figure \(\PageIndex{30}\) shows an interactive iCn3D model of a solution conformation of a parallel DNA triple helix (1BWG).
.png?revision=1&size=bestfit&height=315)
Triple helix formation can also occur within a single strand of DNA. The resulting structure is called H-DNA. An example is shown below. Note that the central blue, black, and red sequences are all mirror-image repeats (around a central nucleotide). During processes that unravel DNA (replication, transcription, repair), the self-association of individual mirror repeats can form a locally stable triple helix, as shown in Figure \(\PageIndex{31}\).

Figure \(\PageIndex{31}\): Schematic illustrations of (A) the H-DNA or intramolecular triplex structure used in this study; del Mundo et al. (2019) Nucleic acids research. 47. e73. 10.1093/nar/gkz237. Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/)
The * between in the G*G and A*A denote Hoogsteen hydrogen bonding (purine motifs) in this intramolecular triple helix. Reverse Hoogsteen hydrogen bonds can also occur.
Triple helices can develop when single-stranded DNA, formed during replication, transcription, or DNA repair with half of the required mirror symmetry, folds back into the adjacent major groove and base pairs, using Hoogsteen/reverse Hoogsteen bonding, which Mg2+ can stabilize.
Recent Updates: Four-Way Junctions
As we will see in the next section on RNA, nonhelical sections of DNA can bind small target molecules through noncovalent interactions. (RNA examples that we will see in the next chapter section include aptamers, ribozymes, and riboswitches.) One example is "lettuce" single-stranded DNA that can bind small fluorophores modeled after the intrinsic fluorophore of the green fluorescent protein. The fluorophore's fluorescence is dramatically enhanced when bound to the lettuce DNA. Figure \(\PageIndex{32}\) below shows the structure of extrinsic DNA fluorophores based on GFP that bind to the single-stranded "lettuce" DNA.

Figure \(\PageIndex{32}\): Structure of extrinsic DNA fluorophores based on GFP that bind to DNA. The font color of the names indicates the color of the emitted fluorescence.
Figure \(\PageIndex{33}\) shows an interactive iCn3D model of a solution conformation of a ssDNA:DFAME fluorophore complex (8FI0). The blue dotted lines show π-π stacking interactions, and the green dotted line a hydrogen bond.
.png?revision=1)
Figure \(\PageIndex{33}\): Solution conformation of a ss-53 mer DNA:DFAME fluorophore complex (8FI0). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...97eLqwNWmaTNC9
The DFAME ligand is shown as sticks.
Figure \(\PageIndex{34}\) shows a close-up of DFAME (colored spacefill) bound to the lettuce DNA.
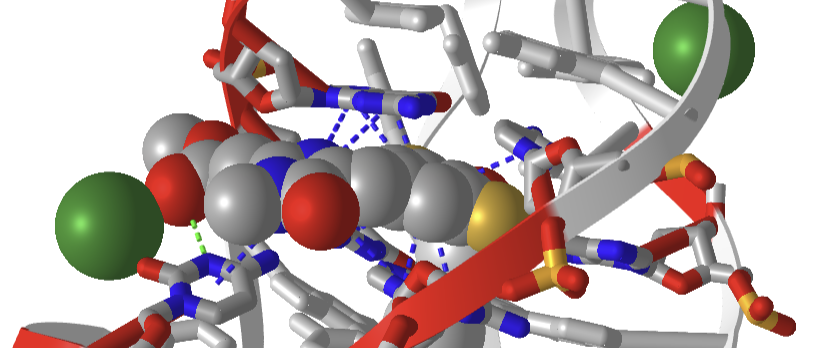
Figure \(\PageIndex{34}\): Pi stacking interactions (blue dotted lines) between the extrinsic fluorophore DFAME (spacefill) and ssDNA.
The DNA fold is characterized as a four-way junction (also seen in RNA, but they are more L or H-shaped). On either end are B-DNA duplexes, and the ssDNA between them forms stem-loops with odd base pairings in the stems. The overall structure is like a cloverleaf. Two coaxial stacks of nucleotides form a G-quadruplex where the fluorophore binds. Pi base stacking between diagonally packed bases and the binding of Mg2+ and K+ stabilize the structure.
Stability of nucleic acids
After looking at the myriad of structures showing the nearly parallel hydrogen-bonded base pairs and from ideas from most textbooks and classes you have taken, you probably think that double-stranded DNA is held together and stabilized by hydrogen bonds between the bases. It is well known that the greater the percentage of GC compared to AT, the greater the stability of the dsDNA. This translates into a higher "melting temperature (TM)," the temperature at which the dsDNA is converted to ssDNA. There is a linear relationship between GC content and TM. The figures above show that GC base pairs have three inter-base hydrogen bonds compared to 2 in AT base pairs. These observations support the notion that inter-base hydrogen bonds are the source of dsDNA stability.
You would be, in general, correct in this belief. Still, you'd be missing the more important contributor to ds-DNA stability, base (π) stacking, and the noncovalent interactions associated with the stacking. The main contributors to stability are hydrophobic interactions in the anhydrous hydrogen-bonded base pairs in the helix. Given that the hydrogen bond donors and acceptors that contribute to base pairing exist in the absence of competing water, the donors and acceptors are free to engage in bonding fully. The hydrogen bond interaction energy is more favorable in the stack. The stacking energy is similar for an AT-AT stack and a GC-GC stack (about -9.8 kcal/mol, 41 kJ/mol). Hence, the AT and GC base pairs contribute equally to stability. The excess stability of dsDNA enriched in GC base pairs can still be explained by the extra stabilization for an additional hydrogen bond per GC base pair
A myriad of interactions stabilizes proteins, but the folded state is marginally more stable than the ensemble of the unfolded state. Marginal stability is important as protein conformation often must be perturbed by binding and ensuing functions. The same must be true of double-stranded DNA, which must "unfold' or separate on replication, transcription, and repair. It is well known that dsDNA structure is sensitive to hydration (see the section on A, B, and Z DNA). As we saw with proteins, small molecules like urea can also denature DNA into single strands.
DNA must be stable enough to carry genetic information but dynamic enough to allow events that require partial unfolding. Other water-soluble molecules like ethylene glycol ethers (polyethylene glycol-400) and diglyme (dimethyl ether of diethylene glycol), which are more hydrophobic than water, appear to reduce base stacking interactions while maintaining them and, at the same time, allow a longitudinal extension or breathing of the helix. This dynamic extension may be required for transitions of B-DNA to Z-DNA, for example. The extensions also allow transient "holes" to appear between base pairs, which might assist in binding intercalating agents like some transition metal complexes. The extension caused by these ethers and natural extensions would decrease base stacking but also strengthen the hydrogen bonding between bases.
Longitudinal helical extensions might be important when homologous genes recombine. In that process, the homologous DNA strand is exchanged with a paired homolog. This processing is associated with strand extension and disruption of base pairs at every third base. Recombination also must allow chain extension as it maintains base-pairing fidelity.
DNA structures get more complicated as they pack into the nucleus of a cell and form chromosomes, as shown in Figure \(\PageIndex{35}\). We will study the packing of DNA in other sections.
 Figure \(\PageIndex{35}\): Packing of DNA into the chromosome. https://commons.wikimedia.org/wiki/F...omosome_en.svg. Creative Commons Attribution 3.0 Unported
Figure \(\PageIndex{35}\): Packing of DNA into the chromosome. https://commons.wikimedia.org/wiki/F...omosome_en.svg. Creative Commons Attribution 3.0 UnportedSummary
This chapter provides an in-depth overview of nucleic acids, one of the four fundamental classes of biological macromolecules, emphasizing their central role in storing and transmitting genetic information. It begins by contrasting DNA and RNA in terms of their chemical composition—highlighting that DNA contains deoxyribose and the bases adenine, guanine, cytosine, and thymine, while RNA uses ribose and replaces thymine with uracil. The discussion covers the nucleoside and nucleotide building blocks and explains how the sugar-phosphate backbone is constructed through the formation of phosphodiester bonds, emphasizing the directionality of nucleic acid synthesis (5′ to 3′).
The chapter then explores the structural organization of DNA and RNA. DNA’s iconic double helix, stabilized by Watson–Crick base pairing and base-stacking interactions, is examined along with its major and minor grooves, which serve as critical recognition sites for proteins. In contrast, RNA, though generally single-stranded, adopts complex secondary and tertiary structures that enable diverse functions beyond mere information storage.
Additionally, alternative base pairing modes—including reverse Watson–Crick, wobble, Hoogsteen, and reverse Hoogsteen interactions—are discussed, illustrating how nucleic acids can form noncanonical structures that influence replication, transcription, and repair. The synthesis and dynamic behavior of nucleic acids are also addressed, including the mechanism of nucleotide incorporation by polymerases and the energetic contributions from pyrophosphate hydrolysis. Finally, the chapter contextualizes these structural features within the historical framework of nucleic acid research, noting landmark discoveries and the evolution of our understanding of DNA and RNA structures.
This comprehensive review lays the foundation for further exploration into the functional roles of nucleic acids in cellular processes, highlighting both the simplicity of their repeating structures and the complexity arising from their dynamic conformations and interactions.
References
Börner, R., Kowerko, D., Miserachs, H.G., Shaffer, M., and Sigel, R.K.O. (2016) Metal ion induced heterogeneity in RNA folding studied by smFRET. Coordination Chemistry Reviews 327 DOI: 10.1016/j.ccr.2016.06.002 Available at: https://www.researchgate.net/publication/303846502_Metal_ion_induced_heterogeneity_in_RNA_folding_studied_by_smFRET
Hardison, R. (2019) B-Form, A-Form, and Z-Form of DNA. Chapter in: R. Hardison’s Working with Molecular Genetics. Published by LibreTexts. Available at: https://bio.libretexts.org/Bookshelves/Genetics/Book%3A_Working_with_Molecular_Genetics_(Hardison)/Unit_I%3A_Genes%2C_Nucleic_Acids%2C_Genomes_and_Chromosomes/2%3A_Structures_of_Nucleic_Acids/2.5%3A_B-Form%2C_A-Form%2C_and_Z-Form_of_DNA
Lenglet, G., David-Cordonnier, M-H., (2010) DNA-destabilizing agents as an alternative approach for targeting DNA: Mechanisms of action and cellular consequences. Journal of Nucleic Acids 2010, Article ID: 290935, DOI: 10.4061/2010/290935 Available at: https://www.hindawi.com/journals/jna/2010/290935/
Mechanobiology Institute (2018) What are chromosomes and chromosome territories? Produced by the National University of Singapore. Available at: https://www.mechanobio.info/genome-regulation/what-are-chromosomes-and-chromosome-territories/
National Human Genome Research Institute (2019) The Human Genome Project. National Institutes of Health. Available at: https://www.genome.gov/human-genome-project
Wikipedia contributors. (2019, July 8). DNA. In Wikipedia, The Free Encyclopedia. Retrieved 02:41, July 22, 2019, from https://en.Wikipedia.org/w/index.php?title=DNA&oldid=905364161
Wikipedia contributors. (2019, July 22). Chromosome. In Wikipedia, The Free Encyclopedia. Retrieved 15:18, July 23, 2019, from en.Wikipedia.org/w/index.php?title=Chromosome&oldid=907355235
Wikilectures. Prokaryotic Chromosomes (2017) In MediaWiki, Available at: https://www.wikilectures.eu/w/Prokaryotic_Chromosomes
Wikipedia contributors. (2019, May 15). DNA supercoil. In Wikipedia, The Free Encyclopedia. Retrieved 19:40, July 25, 2019, from en.Wikipedia.org/w/index.php?title=DNA_supercoil&oldid=897160342
Wikipedia contributors. (2019, July 23). Histone. In Wikipedia, The Free Encyclopedia. Retrieved 16:19, July 26, 2019, from en.Wikipedia.org/w/index.php?title=Histone&oldid=907472227
Wikipedia contributors. (2019, July 17). Nucleosome. In Wikipedia, The Free Encyclopedia. Retrieved 17:17, July 26, 2019, from en.Wikipedia.org/w/index.php?title=Nucleosome&oldid=906654745
Wikipedia contributors. (2019, July 26). Human genome. In Wikipedia, The Free Encyclopedia. Retrieved 06:12, July 27, 2019, from en.Wikipedia.org/w/index.php?title=Human_genome&oldid=908031878
Wikipedia contributors. (2019, July 19). Gene structure. In Wikipedia, The Free Encyclopedia. Retrieved 06:16, July 27, 2019, from en.Wikipedia.org/w/index.php?title=Gene_structure&oldid=906938498