
12.7: Chronic Myelogenous Leukemia (CML)
- Page ID
- 5088

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Leukemia is an uncontrolled proliferation of one kind of white blood cell (or leukocyte). Like all cancers (probably), all the leukemic cells are descended from a single cell that lost the ability to maintain normal control over the cell cycle. There are a number of types of leukemia, as you would expect from the number of types of white blood cells (5) and the number of stages they pass through as they mature. One of the most common is chronic myelogenous leukemia or CML.
Chronic Myelogenous Leukemia (CML) arises in a bone marrow stem cell that is the precursor to all the types of blood cells. However, it usually affects the so-called myeloid lineage (hence the name) that produces granulocytes and macrophages. As the name suggests, the disease often exists for years with only moderately elevated numbers of leukemic cells (descended from the stem cells) and few symptoms. At some point, however, the patient goes through a "blast crisis" when the leukemic granulocyte-macrophage progenitors begin to divide by themselves — increasing their numbers enormously while failing to continue their differentiation.
The Philadelphia Chromosome (Ph1)
In most cases of CML, the leukemic cells share a chromosome abnormality not found in any nonleukemic white blood cells, nor in any other cells of the patient's body. This abnormality is a reciprocal translocation between one chromosome 9 and one chromosome 22. This translocation is designated t(9;22). It results in one chromosome 9 longer than normal and one chromosome 22 shorter than normal. The latter is called the Philadelphia chromosome and designated \(Ph^1\).
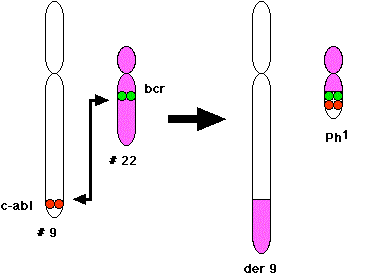
The DNA removed from chromosome 9 contains most of the proto-oncogene designated c-ABL. The break in chromosome 22 occurs in the middle of a gene designated BCR. The resulting Philadelphia chromosome has the 5' section of BCR fused with most of c-ABL.
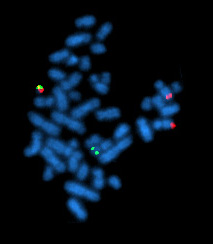
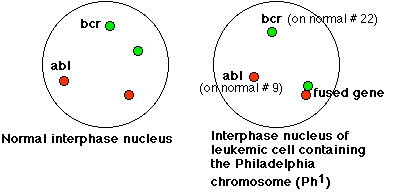
The micrograph in Figure \(\PageIndex{2}\) uses fluorescence in situ hybridization (FISH) to reveal the ABL DNA (red) and the BCR DNA (green) in the interphase nuclei of the leukemic cells of a patient with CML. The red dot at left center reveals the location of ABL on the normal chromosome 9; the green dot (top center) shows BCR on the normal chromosome 22. The combined dots (red + green = yellow) at the lower right reveal the fused BCR-ABL gene on the Philadelphia chromosome. Figure 12.7.3 is a schematic which can help you interpret the micrograph.
Transcription and translation of the hybrid BCR-ABL gene produces an abnormal ("fusion") protein that activates constitutively (all the time) a number of cell activities that normally are turned on only when the cell is stimulated by a growth factor, such as platelet-derived growth factor (PDGF).
This unrestrained activation increases the rate of mitosis and protects the cell from apoptosis. The outcome is an increase in the number of Ph1-containing cells. During the chronic phase of the disease, these are still able to exit the cell cycle and to differentiate into mature cells that perform their normal functions. At some point, however, another mutation in a proto-oncogene (RAS, for example) or in a tumor-suppressor gene (p53, for example), will occur in one of these cells. The additional mutation causes the rate of mitosis in that cell and its descendants to rise sharply. The daughter cells fail to differentiate and the patient enters the crisis phase of the disease.
Until recently, the only successful treatment of CML was to destroy the patient's bone marrow and then restore blood-cell production by infusing stem cells from the bone marrow of a healthy donor. But now treatment with the drug imatinib mesylate (Gleevec® also known STI571) appears to be able to cure the disease. This molecule fits into the active site of the ABL protein preventing ATP from binding there. Without ATP as a phosphate donor, the ABL protein cannot phosphorylate its substrate(s). A phase 2 study, found that almost 90% of the CML patients treated with the drug showed no further progression of their disease.
Gleevec also shows promise against one type of stomach cancer (gastrointestinal stromal tumors = GIST), which is a life-threatening excessive production of eosinophils. In this disease, Gleevec inhibits a different overactive tyrosine kinase. This one also results from the fusion of parts two different genes (because of the deletion of the DNA between them):
- the first 233 codons of a gene designated FIP1L1 fused to
- the final 523 codons of the gene (PDGFRα) encoding the tyrosine kinase domain of a receptor for platelet-derived growth factor. The fusion protein produced, like BCR-ABL, is hyperactive.