
Polymerases
- Page ID
- 324

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Of all the enzymatic functions needed for replication of DNA, the ability to catalyze the incorporation of deoxynucleotides into DNA is most central. Enzymes that catalyze this reaction, DNA polymerases, have been isolated from many species, and many species have multiple DNA polymerases. Our earliest and most complete understanding of the mechanism of these enzymes comes from studies of the first DNA polymerase isolated, called DNA polymerase I.
Mechanism of nucleotide addition by DNA polymerases
In 1956 Arthur Kornberg and his co-workers isolated a protein from E. colithat has many of the properties expected for a DNA polymerase used in replication. In particular, it catalyzes synthesis of DNA from deoxynucleotides, it requires a template and it synthesizes the complement of the template. It is a single polypeptide chain of 928 amino acids, and it is the product of the polAgene. We now understand that this an abundant polymerase, but rather than synthesizing new DNA at the replication fork, it is used during the process of joining Okazaki fragments after synthesis and in DNA repair. Detailed studies of DNA polymerase I have been invaluable to our understanding of the mechanisms of polymerization. Although DNA polymerase I is not the replicative polymerase in E. coli, homologous enzymes are used in replication in other species. Also, the story of how the replicative DNA polymerases were detected in E. coliis a classic illustration of the power of combining biochemistry and genetics to achieve a more complete understanding of an important cellular process.
DNA polymerase I catalyzes the polymerization of dNTPs into DNA. This occurs by the addition of a dNTP (as dNMP) to the 3' end of a DNA chain, hence chain growth occurs in a 5' to 3' direction (Figure 5.11). In this reaction, the 3' hydroxyl at the end of the growing chain is a nucleophile, attacking the phosphorus atom in the a-phosphate of the incoming dNTP. The reaction proceeds by forming a phosphoester between the 3' end of the growing chain and the 5' phosphate of the incoming nucleotide, forming a phosphodiester linkage with the new nucleotide and liberating pyrophosphate (abbreviated PPi). Thus in this reaction, a phosphoanhydride bond in the dNTP is broken, and a phosphodiester is formed. The free energy change for breaking and forming these covalent bonds is slightly unfavorable for the reaction as shown. However, additional noncovalent interactions, such as hydrogen bonding of the new nucleotide to its complementary nucleotide and base-stacking interactions with neighboring nucleotides, contribute to make a total free energy change that is favorable to the reaction in the synthetic direction. Nevertheless, at high concentrations of pyrophosphate, the reaction can be reversed. In the reaction in the reverse direction, nucleotides are progressively removed and released as dNTP in a pyrophosphorolysisreaction. This is unlikely to be of large physiological significance, because a ubiquitous pyrophosphatase catalyzes the hydrolysis of the pyrophosphate to molecules of phosphate. This latter reaction is strongly favored thermodynamically in the direction of hydrolysis. Thus the combined reactions of adding a new nucleotide to a growing DNA chain and pyrophosphate hydrolysis insure that the overall reactions favors DNA synthesis. The basic chemistry of addition of nucleotides to a growing polynucleotide chain outlined in Figure 5.11 is common to virtually all DNA and RNA polymerases.

The DNA synthesis reaction catalyzed by DNA polymerase I requires Mg2+, which is a cofactor for catalysis, and the four deoxynucleoside triphosphates (dNTPs), which are the monomeric building blocks for the growing polymer. The reaction also requires a template strand of DNA to direct synthesis of the new strand, as predicted by the double helical model for DNA and confirmed by the Meselson and Stahl experiment.
This reaction also requires a primer, which is a molecule (usually a chain of DNA or RNA) that provides the 3’ hydroxyl to which the incoming nucleotide is added. DNA polymerases cannot start synthesis on a template by simply joining two nucleotides. Instead, they catalyze the addition of a dNTP to a pre-existing chain of nucleotides; this previously synthesized chain is the primer. The primer is complementary to the template, and the 3’ end of the primer binds to the enzyme at the active site for polymerization (Figure 5.12). When a new DNA chain is being made, once a new nucleotide has been added to the growing chain, its 3' hydroxyl is now the end of the primer. The polymerase moves forward one nucleotide so that this new primer end is at the active site for polymerization. The alternative view, that the DNA primer-template moves while the DNA polymerase remains fixed, is also possible. In both cases the last nucleotide added is now the 3' end of the primer, and the next nucleotide on the template is ready to direct binding of another nucleoside triphosphate.
For the initial synthesis of the beginning of a new DNA chain, a primer has to be generated by a different enzyme; this will be discussed in more detail later in the chapter. For example, short oligoribonucleotides are the primers for the Okazaki fragments; these are found at the 5' ends of the Okazaki fragments and are made by an enzyme called primase. The RNA primers are removed and replaced with DNA (by DNA polymerase I) before ligation.
These requirements for Mg2+, deoxynucleotides and two types of DNA strands (template and primer) were discovered in studies of DNA polymerase I. We now realize that they are also required by all DNA polymerases.
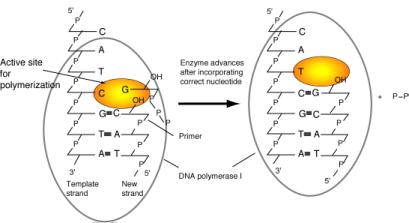
The polymerization active site for DNA polymerase I has a specific dNTP-binding site (Figure 5.12), and the active site adjusts to the deoxynucleotide on the template strand to favor binding of the complementary deoxynucleotide at the active site. Thus the polymerase catalyzes addition to the growing chain of the deoxynucleotide complementary to the deoxynucleotide in the template strand.
In the reaction catalyzed by DNA polymerase I, and all other DNA polymerases studied, the incoming deoxynucleotide is activated. The phosphoanhydride bonds in the triphosphate form of the deoxynucleotide are high-energy bonds (i.e., they have a negative, or favored, free energy of hydrolysis), and the b- and g- phosphates make a good leaving group (as pyrophosphate) after the nucleophilic attack. In contrast, the end of the growing DNA chain is not activated; it is a simple 3'-hydroxyl on the last deoxynucleotide added. This addition of an activated monomer to an unactivated growing polymer is called a tail-growth mechanism. DNA polymerases using this mechanism can only synthesize in a 5' to 3' direction, and all known DNA and RNA polymerases do this. Some other macromolecules, such as proteins, are made by a head-growth mechanism. In this case, the nonactivated end of a monomer attacks the activated end of the polymer. The lengthened chain again contains an activated head (from the last monomer added).
Exercise
Question 5.4. Describe a hypothetical head-growth mechanism for DNA synthesis. In which direction does chain synthesis occur in this mechanism?
Proofreading the newly synthesized DNA by a 3’ to 5’ exonuclease that is part of the DNA polymerase
The protein DNA polymerase I has additional enzymatic activities related to DNA synthesis. One, a 3’ to 5’ exonuclease, is intimately involved in the accuracy of replication. Nucleases are enzymes that catalyze the breakdown of DNA or RNA into smaller fragments and/or nucleotides. An exonuclease catalyzes cleavage of nucleotides from the end of a DNA or RNA polymer. An endonuclease catalyzes cutting within a DNA or RNA polymer. These two activities can be distinguished by the ability of an endonuclease, but not an exonuclease, to cut a circular substrate. A 3’ to 5’ exonuclease removes nucleotides from the 3’ end of a DNA or RNA molecule.
DNA synthesis must be highly accurate to insure that the genetic information is passed on to progeny largely unaltered. Bacteria such as E. colican have a mutation rate, as low as one nucleotide substitution in about 109 to 1010 nucleotides. This low error frequency is accomplished by a strong preference of the polymerase for the nucleotide complementary to the template, which allows about one substitution every 104 to 105 nucleotides. The accuracy of DNA synthesis is enhanced by a proofreading function in the polymerase that removes incorrectly incorporated nucleotides at the end of the growing chain. With proofreading, the accuracy of DNA synthesis is improved by a factor of 102 to 103, so the combined effects of nucleotide discrimination at the polymerization active site plus proofreading allows only about one substitution in 106 to 108 nucleotides. Further reduction in the error rate is achieved by mismatch repair (Chapter 7).
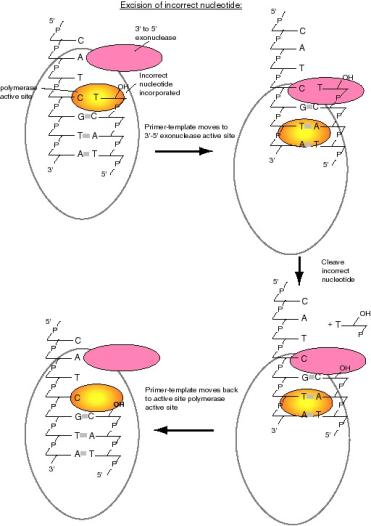
The proofreading function of DNA polymerase I is carried out by a 3' to 5' exonuclease (Figure 5.13). It is located in a different region of the enzyme from the active site for polymerization. When an incorrect nucleotide is added to the 3' end of a growing chain, the rate of polymerization decreases greatly. The primer-template moves to a different active site on the enzyme, the one with the 3’ to 5’ exonucleolytic activity. The incorrect nucleotide is cleaved, and the primer-template moves back to the polymerization active site to resume synthesis.The enzyme distinguishes between correct and incorrect nucleotides at the 3’ end of the primer, such that the 3' to 5' exonuclease much more active when the terminus of the growing chain is not base paired correctly, but the polymerase activity exceeds that of the 3' to 5' exonuclease activity when the correct nucleotide is added.
The polymerizing activity and the proofreading 3' to 5' exonuclease found in DNA polymerase I are also found in most other DNA polymerases. These are central activities to DNA replication.
Tail growth mechanisms allow proofreading and subsequent elongation. If the end of the growing chain were activated (as in head growth), then proofreading would eliminate the activated end and elongation could not continue.
|
|
|
|
Exercise
Question 5.4 Removal of a nucleotide from the 3’ end of the growing chain by a 3’ to 5’ exonuclease is not the reverse of the polymerase reaction. Can you state what the difference is?
Removal of nucleotides by a 5' to 3' exonuclease that is part of DNA polymerase I
In addition to the polymerase and 3’ to 5’ exonuclease common to most DNA polymerases, DNA polymerase I has an unusual 5' to 3' exonucleolytic activity. This enzyme catalyzes the removal of nucleotides in base-paired regions and can excise either DNA or RNA. It is used by the cell to remove RNA primers from Okazaki fragments and in repair of damaged DNA.
This 5' to 3' exonuclease, in combination with the polymerase, has useful applications in the laboratory. One common use is to label DNA in vitro by nick translation (Figure 5.14). In this process, DNA polymerase I will remove the DNA from a nicked strand by the 5' to 3' exonuclease, and then use the exposed 3' hydroxyl at the nick as a primer for new DNA synthesis by the 5' to 3' polymerase, thereby replacing the old DNA. The result is also a movement, or translation, of the nick from one point on the DNA to another, hence the process is called nick translation. If the reaction is carried out in the presence of one or more radiolabeled deoxynucleoside triphosphates (e.g., [a32P] dNTPs), then the new DNA will be radioactively labeled.
A similar process can be used to repair DNA in a cell. As will be discussed in Chapter 7, specific enzymes recognize a damaged nucleotide and cleave upstream of the damage. One way to remove the damaged DNA and replace it with the correct sequence is with the 5' to 3' exonuclease of DNA polymerase I and accompanying DNA synthesis.
![]()

[a32P]
Figure 5.14.The 5' to 3' exonuclease of DNA polymerase I can be used in nick translation to label DNA in vitro.
Structural domains of DNA polymerase I
Further understanding of the mechanism of the three enzymatic functions of DNA polymerase can be obtained from a study of the three-dimensional (3-D) structure of the protein. Much of our knowledge of the structure of DNA polymerase I has come from biochemical characterization and more recently by determination of the 3-D structure using X-ray crystallography. These studies have shown that distinct structural domains of DNA polymerase I contain the different catalytic activities. Also, the 3-D structure provided the first look at what is now recognized as a common structure for many polymerases.
Mild treatment with the protease subtilisin cleaves DNA polymerase I into two fragments. The small fragment contains the 5' to 3' exonuclease, and the larger, or "Klenow," fragment (named for the biochemist who did the cleavage analysis) contains the polymerase and the proofreading 3' to 5' exonuclease (Figure 5.15). Thus the two activities common to most polymerases are together in the Klenow fragment, whereas the distinctive 5' to 3' exonuclease is in a separable domain. The fact that a mild treatment with a protease without a precise sequence specificity indicates that an exposed, readily cleaved domain connects the large and small fragments. Both these observations suggest that the 5' to 3' exonuclease was an active domain added to a polymerase plus proofreading domain during the evolution of E. coli. The Klenow polymerase is used in several applications in the laboratory, e.g., labeling the ends of restriction fragments by filling in the overhangs and sequencing by the dideoxynucleotide chain termination method.

The 3-D structure of the large fragment of DNA polymerase I, determined by crystallography, provides additional insight into the enzymatic functions of key structural components. The large fragment has a deep cleft, about 30 Å deep, into which the template strand and primer bind. This cleft resembles a "cupped right hand" as illustrated in Figure 5.16. The "palm" is formed by a series of b-sheets and the thumb and fingers are made by a-helices. The polymerase active site has been mapped within the deep cleft, with contributions from the b-sheets that form the palm and the a-helices forming the fingers. You can see more detailed views of the structure of the Klenow fragment at the Course/Book web site (currently www.bmb.psu.edu/courses/bmb400/default.htm. Click on the link to kinetic images, download the MAGE program and the kinemage file for DNA polymerase I, and view them on your own computer.)
The 3' to 5' proofreading exonuclease is located in another part of the structure of the Klenow fragment, about 25 Å from the polymerase active site. Thus the primer terminus has to move this distance in order for the enzyme to remove misincorporated nucleotides.


The large Klenow fragment of the E. coliDNA polymerase I lacks the 5’ to 3’ exonuclease, so the 3-D structure of the Klenow fragment gives no information about that exonuclease. However, the 5’ to 3’ exonuclease domain can be seen in the structure of DNA polymerase from the thermophilic bacterium Thermus aquaticus. This protein structure is very similar to that of DNA polymerase I of E. coliin the polymerase and 3’ to 5’ exonuclease domains, and it has an additional 5' to 3' exonuclease domain located about 70 Å from the polymerase active site. This is a large distance, but remember that this exonuclease is working on a different region of the DNA molecule than the polymerase. The 5’ to 3’ exonuclease uses one part of the DNA molecule as a substrate for excising primers or removing damaged DNA, whereas the polymerase uses a different part of the DNA molecule as a template to direct synthesis of a new strand.
Curiously, a region homologous to the proofreading 3' to 5' exonuclease domain of DNA polymerase I is present in the Thermus aquaticuspolymerase structure, but it is no longer functional. The absence of proofreading accounts for the elevated error rate in this polymerase used very commonly for amplification of DNA by PCR. Of course, this polymerase is used in PCR because it is stable at the high temperatures encountered during the cycles of PCR. Some other thermostable polymerases with a lower error rate have become available more recently for use in PCR.
Similar "cupped right hand" structures occur in the tertiary structure of T7 RNA polymerase and the HIV reverse transcriptase. Thus DNA polymerase I was the first member described in what we now realize is a large class of nucleic acid polymerases. This family includes single unit polymerases for both RNA and DNA synthesis. You can access a tutorial on the T7 DNA polymerase at www.clunet.edu/BioDev/omm/exh...s.htm#displays. This structure has some similarities to that of DNA polymerase I.
Physiological role of DNA polymerase I
Although studies of DNA polymerase I have provided much information about the mechanism of DNA synthesis, genetic analysis has shown that the polymerase function of this enzyme is not required for DNA replication. DNA polymerase I is encoded by the polAgene in E. coli.However, no mutant allele of polAwas isolated in screens for conditional mutants defective in DNA replication. The most compelling argument that this polymerase is not required for replication came from an examination of thousands of E. colimutants, assaying them for DNA polymerase I activity. A mutant polA strain was isolated (Figure 5.16). This mutant allele, called polA1, contained a nonsense codon, leading to premature termination of synthesis of the product polypeptide and hence a loss of polymerase function. However, the mutant strain grew at a normal rate, which shows that DNA polymerase I is notrequired for DNA synthesis. The most striking phenotype of the polA1mutant was its strongly reduced ability to repair DNA damage. Further investigation led to the isolation of conditional lethal alleles of the polAgene. The mutant DNA polymerase I proteins encoded by these conditional lethal alleles are defective in the 5' to 3' exonuclease activity, demonstrating that this activity is required for cell viability. The 5' to 3' exonuclease activity removes RNA primers during synthesis of the lagging strand at the replication fork, and it is used in DNA repair.

DNA polymerase III is a highly processive, replicative polymerase
The conclusion that DNA polymerase I is not the replicative polymerase for E. coliled to the obvious question of what enzyme is actually used during replication. Investigation of the genes isolated in screens for mutants that are conditionally deficient in replication led to the answer. The replicative polymerase in E. coliis DNA polymerase III.
DNA polymerase I is more abundant than other polymerases in E. coliand obscures their activity. Thus the depletion of DNA polymerase I activity in polA1mutant cells (Figure 5.17) provided the opportunity to observe the other DNA polymerases. DNA polymerases II and III were isolated from extracts of polA1cells, named in the order of their discovery.
DNA polymerase IIis a single polypeptide chain whose function is uncertain. Strains having a mutated gene for DNA polymerase II (polB1) show no defect in growth or replication. However, the activity of DNA polymerase II is increased during induced repair of DNA, and it may function to synthesize DNA opposite a deleted base on the template strand.
Genetic evidence clearly shows that DNA polymerase IIIis used to replicate the E. coli chromosome. This enzyme is composed of multiple polypeptide subunits. Several of the genes encoding these polypeptide subunits were identified in screens for conditional lethal mutants defective in DNA replication. Loss of function of these dnagenes blocks replication, showing that their products are required for replication.
Low abundance and high processivity of DNA polymerase III
DNA polymerase III has many of the properties expected for a replicative polymerase. One of the complications to studies of DNA polymerase III is that different forms were isolated by various procedures. We now realize that these forms differ in the number of subunits present in the isolated enzyme. For enzymes with multiple subunits, we refer to the complex with all the subunits needed for its major function as the holoenzyme or holocomplex. The DNA polymerase holoenzyme has ten subunits, which will be discussed in detail in the next section.
It is the DNA polymerase holoenzyme that has the properties expected for a replicative polymerase, whereas DNA polymerase I does not (see comparison in Table 5.1). It is less abundantthan DNA polymerase I, but large number of replicative DNA polymerases are not needed in the cell. Only one or two polymerases can be used at each replication fork, so the 10 molecules of the DNA polymerase III holoenzyme will suffice. DNA polymerase III catalyzes DNA synthesis at a considerably higher ratethan DNA polymerase I, by a factor of about 70. The elongation rate measured for the DNA polymerase III holoenzyme (42,000 nucleotides per min) is close to the rate of replication fork movement measured in vivoin E. coli(60,000 nucleotides per min).
A key property for a replicative DNA polymerase is high processivity, which is a striking characteristic of the DNA polymerase III holoenzyme. Processivity is the amount of polymerization catalyzed by an enzyme each time it binds to an appropriate template, or primer-template in the case of DNA polymerases. It is measured in nucleotides polymerized per binding event. In order to replicate the 4.5 megabase chromosome of E. coliin 30 to 40 min, DNA polymerase needs to synthesize DNA rapidly, and in a highly processive manner. DNA polymerase I synthesizes less than 200 nucleotides per binding event, but as the holoenzyme, DNA polymerase III is much more processive, exceeding the limits of the assay used to obtain the results summarized in Table 5.1. In contrast, the DNA polymerase III core, which has only three subunits (see next section), has very low processivity.
Table 5.1. Comparison of DNA polymerases I and III (Pol I and Pol III)
|
Property |
Pol I |
Pol III core |
Pol III holoenzyme |
|---|---|---|---|
|
molecules per cell |
400 |
40 |
10 |
|
nucleotides polymerized min-1 (molecule enzyme)-1 |
600 |
9000 |
42,000 |
|
processivity [nucleotides polymerized per initiation] |
3-188 |
10 |
>105 |
|
5' to 3' polymerase |
+ |
+ |
+ |
|
3' to 5' exonuclease, proofreading |
+ |
+ |
+ |
|
5' to 3' exonuclease |
+ |
- |
- |
Note: + and – refer to the presence or absence of the stated activity in the enzyme.
Question 5.6. If the rate of replication fork movement measured in vivo in E. coliis 60,000 nucleotides per min, how many forks are needed to replicate the chromosome in 40 min? Recall that the size of the E. colichromosome is 4.64 ´ 106 bp.
Subunits and mechanism of DNA polymerase III
The DNA polymerase III enzyme has four distinct functional components, and several of these contain multiple subunits, as listed in Table 5.2 and illustrated in Figure 5.18. The a and e subunits contain the major polymerizing and proofreading activities, respectively. They combine with the q subunit to form the catalytic core of the polymerase. This core can be dimerized by the t2 linker protein to form a subassembly called DNA polymerase III'. Addition of the third functional component, the g complex, generates another subassembly denoted DNA polymerase III*. All of these subassemblies have been isolated from E. coliand have been characterized extensively. The final component is the b2 dimer, which when combined with DNA polymerase III* forms the holoenzyme.

The various activities of DNA polymerase III can be assigned to individual subunits (Table 5.2). For instance, the major polymerase is in the a subunit, which is encoded by the dnaEgene (also known as polC). The 3' to 5' exonuclease is in the e subunit, which is encoded by the dnaQgene (also known as the mutDgene). However, maximal activity is obtained with combinations of subunits. The DNA polymerase III core is a complex of the a, e and q subunits, and the activity of the core in both polymerase and 3' to 5' exonuclease assays is higher in than in the isolated subunits.
Table 5.2. Subassemblies of DNA polymerase III, major subunits, genes and functions
|
Functional component |
Subunit |
Mass (kDa) |
Gene |
Activity or function |
|---|---|---|---|---|
|
Core polymerase |
a |
129.9 |
polC=dnaE |
5' to 3' polymerase |
|
e |
27.5 |
dnaQ=mutD |
3'-5' exonuclease |
|
|
q |
8.6 |
Stimulates e exonuclease |
||
|
Linker protein |
t |
71.1 |
dnaX |
Dimerizes cores |
|
Clamp loader |
g |
47.5 |
dnaX |
Binds ATP |
|
(or g complex) |
d |
38.7 |
Binds to b |
|
|
(ATPase) |
d' |
36.9 |
Binds to g and b |
|
|
c |
16.6 |
Binds to SSB |
||
|
y |
15.2 |
Binds to c and g |
||
|
Sliding clamp |
b |
40.6 |
dnaN |
Processivity factor |
The activities of the subunits can be measured in vitroby appropriate biochemical assays. In addition, the phenotype of mutations in the gene encoding a given subunit can show that subunit is required for a particular process. Mutant a subunits are the product of conditional lethal alleles discovered in screens for dnagenes, but they also were discovered as the product of polymerase-defective alleles defining the polCgene. Thus the dnaEgene is the same as same as the polCgene, showing that this subunit with polymerase activity is needed in replication. Similarly, the phenotype of mutations in the gene encoding the e subunit shows that it is needed for proofreading. Mutant alleles of the dnaQgene were identified in a screen for mutator genes, which generate a high frequency of mutants in bacteria when defective. These alleles defined a gene mutD, which was subsequently shown to be the same as dnaQ. The mutator phenotype of mutant dnaQ/mutDstrains results from a lack of proofreading by the e subunit during replication, allowing more frequent incorporation of incorrect nucleotides into DNA.
The b2 dimer is the key protein that confers highprocessivityon DNA polymerase III. Association of the b2 dimer with DNA polymerase III increases the processivity from about 10 nucleotides polymerized per binding event to over 100,000 nucleotides polymerized per binding event (Table 5.1). This dimeric protein forms a ring through which the duplex DNA can pass; the ring will slide easily along DNA unless impeded, as, for example, by proteins bound to the template DNA. Thus the b2 dimer acts as a sliding clamp, holding the polymerase onto the DNA being copied. Once DNA polymerase III is associated with the clamp on DNA, it will polymerize until it reaches the next primer for an Okazaki fragment during lagging strand synthesis. For leading strand synthesis, the DNA polymerase presumably remains associated with the DNA via the b2 clamp until the chromosomal DNA is completely replicated. The 3-D structure of the b2 dimer, determined by X-ray crystallography, shows a macromolecular ring. This structure can be viewed at the web site for the course and at the Online Museum of Macromolecules (www.clunet.edu/BioDev/omm/exh...s.htm#displays).
The g-complex contains several subunits: two molecules of g subunits and one molecule each of d, d', c, and y. It loadsthe b2 dimer clamp onto a primer-template, in a process that requires ATP hydrolysis (Figure 5.19). The catalytic core of DNA polymerase III will then link to the template-bound clamp and will initiate highly processive replication. The g-complex also serves to unload the clamp once an Okazaki fragment is completed during lagging strand synthesis; hence it is both a clamp loader and unloader, allowing the polymerase and the clamp to cycle repeatedly from one Okazaki fragment to another.
The g-complex carries out these opposite activities on different structures, loading on the clamp at a template-primer and unloading the clamp at the end of a completed Okazaki fragment. For instance, encountering the 5' end of the previously synthesized Okazaki fragment may be the distinctive structure that shifts the g-complex into its unloading mode. It does not unload the clamp while DNA polymerase III is catalyzing polymerization.
Figure 5.19 illustrates the proposed steps in this process. The g-complex in the ATP-bound form binds the b2 clamp, whereas the g-complex in the ADP-bound form releases the b2 clamp. Thus loading and unloading depend on a round of ATP hydrolysis. When the g-complex in the ATP-bound form binds the b2 clamp, the DNA polymerase III holoenzyme is in a conformation that allows it to find a primer-template. The ring of the b2 clamp is held open by the g-complex-ATP, allowing it to bind around a primer-template. Hydrolysis of ATP by the g-complex leaves it in an ADP-bound form. In this new, ADP-bound conformation of the g-complex, it dissociates from the b2 clamp, thereby allowing the b2 clamp to bind to the catalytic core of the holoenzyme and also close around the primer-template. The holoenzyme is now ready to catalyze processive DNA synthesis. Elongation continues until the holoenzyme encounters a previously synthesized Okazaki fragment. Now the g-complex binds ATP (presumably by an ADP-ATP exchange reaction) and shifts into the conformation for binding to the b2 clamp and taking it off the DNA template. This half of the holoenzyme is now able to dissociate from the template and find the next primer-template junction to begin synthesis of another Okazaki fragment.
The clamp loading and unloading activities of the g-complex are a cycle of changes in protein associations. These changes occur because of the enzymatic activities of the complex, which in turn alter the conformations of the proteins and their preferred interactions. As shown in Figure 5.19, the g-complex is an ATPase, which is an enzyme that catalyzes the hydrolysis of ATP to ADP and phosphate. It is also an ATP-ADP exchange factor.

Changes in conformation and activity of proteins depending on whether they are bound to a nucleoside triphosphate (ATP or GTP) or a nucleoside diphosphate (ADP or GDP) is a common theme in biochemistry. The GTP-bound forms of proteins, which can be turned off by GTP-hydrolysis and reactivated by GDP-GTP exchange proteins, mediate critical cell signaling events. As will be seen in Chapter 14, GTP- and GDP-bound forms of translation factors carry out opposite functions. Proteins assume different conformations depending on the cofactor bound (in this case a nucleotide), and each conformation has a distinct activity. The ability to change the conformation by a hydrolytic activity (converting ATP to ADP and phosphate) allows the protein to shift activities readily.
The two catalytic cores of DNA polymerase III are joined together by the t subunits to make an asymmetric dimer (see Figure 5.18). The half of the holoenzyme without the g complex is proposed to synthesize the leading strand of new DNA, and the core with the g complex is proposed to synthesize the lagging strand. Both of the cores in the asymmetric dimer are associated with a b2 clamp at the replication fork. In this model, synthesis of boththe leading and lagging strands is catalyzed by the sameDNA polymerase III complex, thereby coordinating synthesis of both new strands strand. Note that if the template for lagging strand synthesis is looped around the enzyme, then leading and lagging strand synthesis would be occurring in the same direction as replication fork movement (Figure 5.20), despite the opposite polarities of the two template strands. Thus the asymmetric dimer model suggests a means to couple both leading strand and lagging strand synthesis.
 Replication machinery
Replication machinery
|
|
|
