
9.3: Cloning and Recombinant Expression
- Page ID
- 14971


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Differentiate Cloning Types:
• Explain the concept of molecular cloning (creating copies of DNA fragments) versus reproductive cloning (creating an identical whole organism). -
Outline the Cloning Workflow:
• Describe the steps required to isolate, purify, amplify, analyze, and sequence target DNA for cloning and subsequent expression studies. -
Understand Cloning Vectors:
• Identify key components of a cloning vector—including the origin of replication, selectable marker, and multiple cloning site (MCS)—and explain their functions in gene cloning. -
Recognize Various Cloning Systems:
• Compare the use of different vectors (e.g., plasmids, bacteriophages, cosmids, BACs, YACs, shuttle vectors, and human artificial chromosomes) and discuss how the choice depends on factors like insert size and host compatibility. -
Describe Restriction Enzymes and Their Function:
• Explain how restriction endonucleases recognize specific palindromic sequences, produce sticky or blunt ends, and are essential tools for generating compatible DNA fragments for ligation. -
Explore Alternative Cloning Methods:
• Discuss techniques such as TOPO cloning and Gateway recombination that allow gene insertion without traditional restriction digestion and ligation steps. -
Apply Selectable and Reporter Markers:
• Describe how selectable markers (e.g., antibiotic resistance genes) and reporter genes (e.g., GFP, lacZα) are used to screen for successful cloning events. -
Connect Cloning to Gene Expression:
• Explain how vectors are engineered with expression elements like promoters and ribosomal binding sites to drive transcription and translation in host cells, and the importance of inducible systems for toxic proteins. -
Discuss Subcloning Techniques:
• Understand the rationale and process of subcloning—transferring a DNA fragment from one vector to another—to tailor vectors for specific downstream applications. -
Review Recombinant DNA and Protein Production:
• Define recombinant DNA molecules and recombinant proteins, and describe how cloning techniques enable the production and study of these molecules. -
Summarize Reproductive Cloning:
• Outline the method of somatic cell nuclear transfer used in reproductive cloning, using examples such as Dolly the sheep, and discuss its challenges and implications. -
Examine Genetic Engineering Applications:
• Describe how recombinant DNA technology is used to create genetically modified organisms (GMOs) and transgenic organisms, and highlight the differences between reverse genetics and classical genetic approaches. -
Understand CRISPR-Cas Technology:
• Explain the natural role of CRISPR in prokaryotic antiviral defense, and describe how the CRISPR-Cas9 system is repurposed for targeted gene editing in various organisms. -
Evaluate the Impact of CRISPR on Biotechnology:
• Discuss the revolutionary applications of CRISPR-Cas9 in research, medicine, and agriculture, including its use for gene knockout, precise genome editing, and therapeutic interventions. -
Integrate Concepts Across Cloning and Gene Editing:
• Compare and contrast traditional molecular cloning techniques with modern gene-editing methods, emphasizing how the ability to manipulate DNA has transformed our understanding of biology and enabled new biotechnological applications.
These learning goals are intended to guide your exploration of the methodologies and applications of DNA cloning and gene editing, linking foundational techniques with cutting-edge advances in biotechnology.
To clone a gene from an organism and express it in either prokaryotic or eukaryotic cells, DNA from a target source must be isolated, purified, amplified, analyzed, and sequenced as described in previous sections.
Cloning
In general, cloning means the creation of a perfect replica. Typically, the word describes the creation of a genetically identical copy. In biology, re-creating a whole organism is called “reproductive cloning.” Long before attempts were made to clone an entire organism, researchers learned how to copy short stretches of DNA—a process called molecular cloning.
Molecular cloning allows for the creation of multiple copies of genes, the expression of genes, and the study of specific genes. The fragment is first inserted into a cloning vector to get the DNA fragment into a bacterial cell in a form that will be copied or expressed.
Cloning vector
A cloning vector is a small piece of DNA that can be stably maintained in an organism, and a foreign DNA fragment can be inserted into it for cloning purposes. The cloning vector may be DNA taken from a virus, a higher organism's cell, or a bacterium's plasmid. The vector contains features that allow for the convenient insertion or removal of a DNA fragment to or from the vector, for example, by treating the vector and the foreign DNA with a restriction enzyme that cuts the DNA. DNA fragments thus generated contain either blunt ends or overhangs known as sticky ends, and vector DNA and foreign DNA with compatible ends can then be joined together by molecular ligation. A DNA fragment cloned into a cloning vector may be further subcloned into another vector designed for more specific use.
There are many types of cloning vectors, but the most commonly used are genetically engineered plasmids. Cloning is generally first performed using Escherichia coli, and cloning vectors in E. coli include plasmids, bacteriophages (such as phage λ), cosmids, and bacterial artificial chromosomes (BACs). However, some DNA, such as very large DNA fragments, cannot be stably maintained in E. coli. Other organisms, such as yeast, may be used for these studies. Cloning vectors in yeast include yeast artificial chromosomes (YACs). The common bacterial cloning plasmid, pRB322, is shown in Figure \(\PageIndex{1}\).
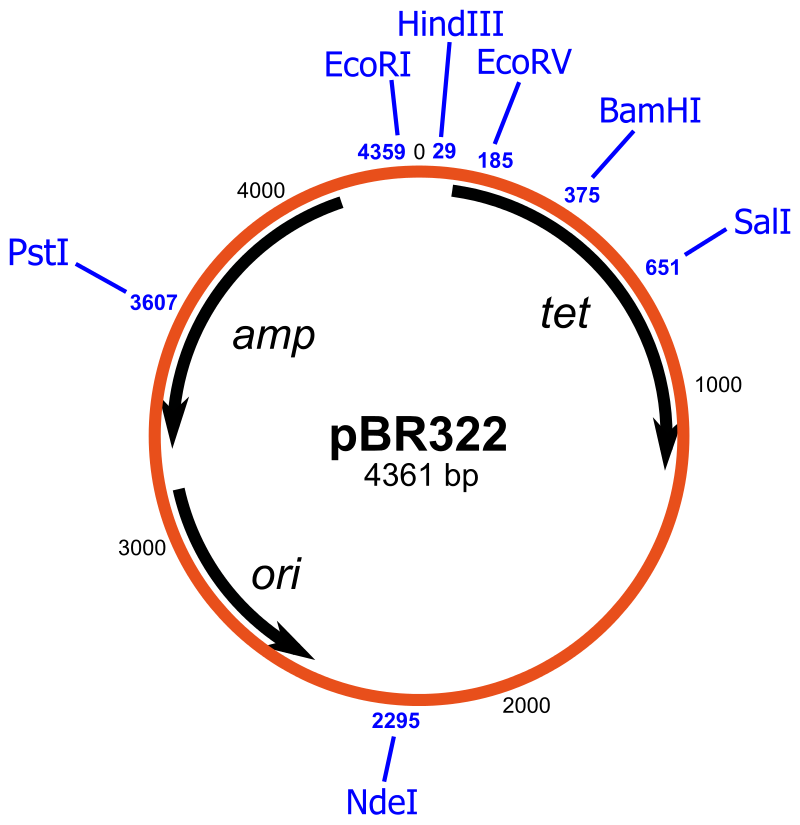
All commonly used cloning vectors in molecular biology have key features necessary for their function, such as a suitable cloning site with restriction enzymes and a selectable marker. Others may have additional features specific to their use. Cloning is often performed using E. coli for ease and convenience. Thus, cloning vectors often have elements necessary for their propagation and maintenance in E. coli, such as a functional origin of replication (ori). The ColE1 origin of replication is found in many plasmids. Some vectors also include elements that allow them to be maintained in another organism in addition to E. coli; these vectors are called shuttle vectors.
Cloning site
All cloning vectors have features that allow a gene to be conveniently inserted or removed from the vector. This may be a multiple cloning site (MCS) or polylinker containing many unique restriction sites. The restriction sites in the MCS are first cleaved by restriction enzymes, and then a PCR-amplified target gene, which is also digested with the same enzymes, is ligated into the vectors using DNA ligase. If desired, the target DNA sequence can be inserted into the vector in a specific direction. The restriction sites may be further sub-cloned into another vector if necessary.
Other cloning vectors may use topoisomerase instead of ligase, and cloning may be done more rapidly without the need for restriction digesting or inserting the vector. In this TOPO cloning method, a linearized vector is activated by attaching topoisomerase I to its ends, and this "TOPO-activated" vector may then accept a PCR product by ligating both the 5' ends of the PCR product, releasing the topoisomerase and forming a circular vector in the process. Another method of cloning without using a DNA digest and ligase is DNA recombination, for example, as used in the Gateway cloning system. Once cloned into the cloning vector (called entry clone in this method), the gene may be conveniently introduced into a variety of expression vectors by recombination.
Restriction Enzymes
Restriction enzymes (restriction endonucleases) recognize and predictably cut specific DNA sequences; bacteria naturally produce them as a defense mechanism against foreign DNA.
As the name implies, restriction endonucleases (or restriction enzymes) are “restricted” in their ability to cut or digest DNA. The restriction that is useful to biochemists is usually a palindromic DNA sequence. Palindromic sequences are the same sequence, forwards and backward. Some examples of palindromes: RACE CAR, CIVIC, A MAN A PLAN A CANAL PANAMA. DNA has two complementary strands. Therefore, the reverse complement of one strand is identical to the other.
Like a palindromic word, the DNA palindromic sequence reads the same forward and backward. The sequence usually reads the same forward on one strand and backward on the complementary strand. Restriction enzymes often cut DNA into a staggered pattern. When a staggered cut is made in a sequence, the overhangs are complementary, as shown in Figure \(\PageIndex{2}\).
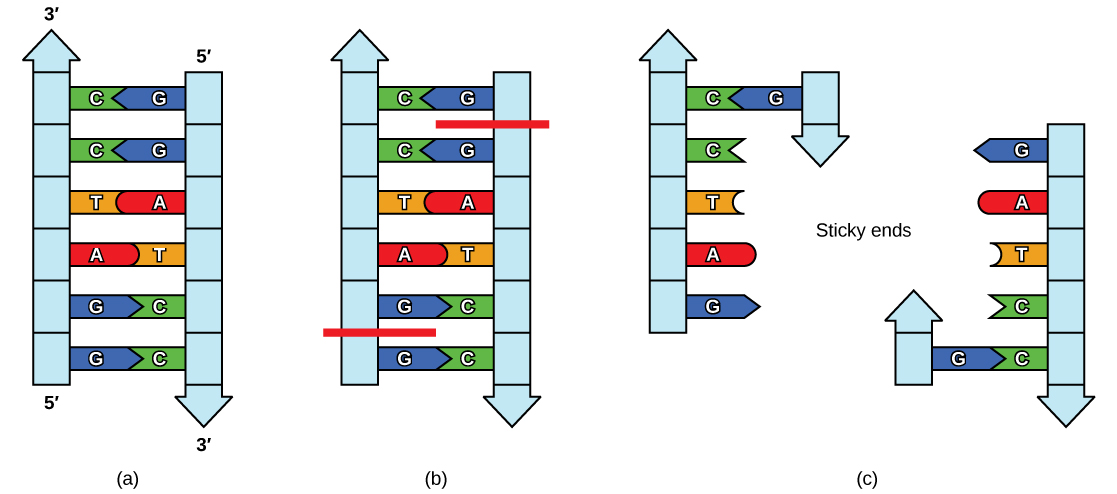
Figure \(\PageIndex{2}\): Restriction Enzyme Recognition Sequences. In this, (a) six-nucleotide restriction enzyme recognition site, notice that the sequence of six nucleotides reads the same in the 5′ to 3′ direction on one strand as it does in the 5′ to 3′ direction on the complementary strand. This is known as a palindrome. (b) The restriction enzyme breaks the DNA strands and (c) the cut in the DNA results in “sticky ends”. Another piece of DNA cut on either end by the same restriction enzyme could attach to these sticky ends and be inserted into the gap made by this cut. http://opentextbc.ca/biology/wp-cont...e_10_01_04.jpg
Molecular biologists also tend to use these special molecular scissors that recognize palindromes of 6 or 8. By using 6-cutters or 8-cutters, the sequences occur rarely but often enough to be useful. Figure \(\PageIndex{3}\) the sequence for HindII cuts.

Figure \(\PageIndex{3}\): Sequence of HindIII stick end cuts.
Figure \(\PageIndex{4}\) shows restriction enzyme cuts that leave sticky or blunt end.


Figure \(\PageIndex{4}\): Restriction Enzymes. Restriction enzymes recognize palindromic sequences in DNA and hydrolyze covalent phosphodiester bonds of the DNA to leave either “sticky/cohesive” or “blunt” ends. This distinction in cutting is important because an EcoRI sticky end can match up a piece of DNA cut with the same enzyme to glue or ligate them back together. While endonucleases cut DNA, ligases join them back together. DNA digested with EcoRI can be ligated back together with another piece of DNA digested with EcoRI, but not to a piece digested with SmaI. Another blunt cutter is EcoRV with a recognition sequence of GAT | ATC.
Selectable marker
The vector carries a selectable marker to allow the selection of positively transformed cells. Antibiotic resistance is often used as a marker, an example being the beta-lactamase gene, which confers resistance to the penicillin group of beta-lactam antibiotics like ampicillin. Some vectors contain two selectable markers. For example, the plasmid pACYC177 has both ampicillin and kanamycin resistance genes. Shuttle vectors designed to be maintained in two organisms may also require two selectable markers. However, some selectable markers, such as resistance to zeocin and hygromycin B, are effective in different cell types. Auxotrophic selection markers that allow an auxotrophic organism to grow in a minimal growth medium may also be used; examples of these are LEU2 and URA3, which are used with their corresponding auxotrophic strains of yeast.
Another selectable marker allows for the positive selection of plasmid with cloned genes. This may involve using a gene lethal to the host cells, such as barnase, Ccda, and the parD/parE toxins. This typically works by disrupting or removing the lethal gene during the cloning process, and unsuccessful clones where the lethal gene still remains intact would kill the host cells. Therefore, only successful clones are selected.
Reporter genes
Reporter genes are used in some cloning vectors to facilitate the screening of successful clones by using features of these genes that allow successful clone clones to be easily identified. Such features present in cloning vectors may be the lacZα fragment for α complementation in the blue-white selection and/or marker gene or reporter genes in frame with and flanking the MCS to facilitate the production of fusion proteins. Examples of fusion partners that may be used for screening are the green fluorescent protein (GFP) and luciferase. Figure \(\PageIndex{5}\) shows such a construct with GFP.

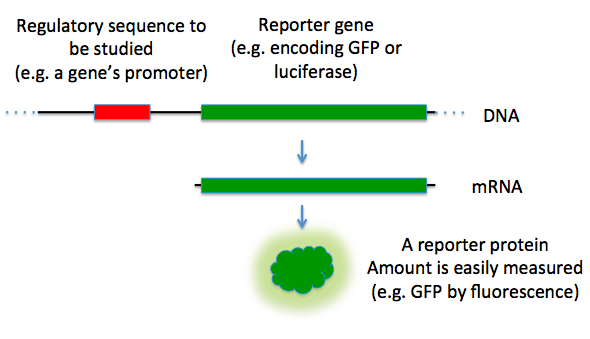
Figure \(\PageIndex{5}\): Reporter Genes. In this diagram, the green fluorescence protein is used as a reporter gene to study upstream regulatory sequences. Image by TransControl
Elements for expression
Suppose the expression of the targeted gene is desired. In that case, a cloning vector also needs to contain suitable elements for expressing the cloned target gene, including a promoter and ribosomal binding site (RBS). The target DNA may be inserted into a site under the control of a particular promoter necessary to express the target gene in the chosen host. Where the promoter is present, the expression of the gene is preferably tightly controlled and inducible so that proteins are only produced when required. Some commonly used promoters are the T7 and lac promoters. The presence of a promoter is necessary when screening techniques such as blue-white selection are used.
Cloning vectors without promoter and RBS for the cloned DNA sequence are sometimes used, for example, when cloning genes whose products are toxic to E. coli cells. Promoter and RBS for the cloned DNA sequence are also unnecessary when making a genomic or cDNA library of clones since the cloned genes are normally subcloned into a more appropriate expression vector if their expression is required.
Types of cloning vectors
Many cloning vectors are available, and choosing the right vector may depend on several factors, such as the insert size, copy number, and cloning method. Large DNA inserts may not be stably maintained in a general cloning vector, especially for those with a high copy number, therefore cloning large fragments may require a more specialized cloning vector.
Plasmids
Plasmids are autonomously replicating circular extra-chromosomal DNA. They are the standard cloning vectors and the ones most commonly used. Most general plasmids may be used to clone DNA inserts to 15 kb in size. Many plasmids have high copy numbers. For example, pUC19 has a copy number of 500-700 copies per cell, and a high copy number is useful as it produces a greater yield of recombinant plasmid for subsequent manipulation. However, low-copy-number plasmids may be preferably used in certain circumstances, such as when the protein from the cloned gene is toxic to the cells.
Bacteriophage
The bacteriophages most commonly used for cloning are the lambda (λ) phage and M13 phage. There is an upper limit on the amount of DNA that can be packed into a phage (a maximum of 53 kb). The average lambda phage genome is roughly 48.5 kb. Therefore, to allow foreign DNA to be inserted into phage DNA, phage cloning vectors may need to have some of their non-essential genes deleted to make room for the foreign DNA. Figure \(\PageIndex{6}\) shows the phage sequence and cartoon structure.
There is also a lower size limit for DNA that can be packed into a phage. This property can be used for selection - vectors without inserts may be too small, therefore only vectors with inserts may be selected for propagation.

Figure \(\PageIndex{6}\): Lambda Phage. (A) Schematic representation of the circular genome of the lambda phage (B) Diagram of the Lambda Phage infectious particle and (C) Electron micrograph of the related bacteriophage, vibriophage VvAWI. The bar denotes 50 nm in length. Images A and C are modified from Nigro, O, Culley, A., and Steward, G.F. (2012) Standards in Genomic Science 6(3):415-26, and image B is from Jack Potte
Cosmids
Cosmids are plasmids that incorporate a segment of bacteriophage λ DNA that has the cohesive end sites (cos), which contain elements required for packaging DNA into λ particles. It is typically used to clone large DNA fragments between 28 and 45 Kb.
Bacterial artificial chromosome
Insert sizes up to 350 kb can be cloned in a bacterial artificial chromosome (BAC). BACs are maintained in E. coli with a copy number of only 1 per cell. BACs have often been used to sequence the genome of organisms in genome projects, including the Human Genome Project. A short piece of the organism's DNA is amplified as an insert in BACs and then sequenced. Finally, the sequenced parts are rearranged in silico, resulting in the organism's genomic sequence. BACs have largely been replaced in this capacity with faster and less laborious sequencing methods like whole genome shotgun sequencing and now, more recently, next-gen sequencing.
Yeast artificial chromosome
Yeast artificial chromosomes are used as vectors to clone DNA fragments of more than 1 megabase (1Mb = 1000kb = 1,000,000 bases) in size. They are useful in cloning larger DNA fragments as required in mapping genomes, such as in the human genome project. It contains a telomeric sequence, an autonomously replicating sequence( features required to replicate linear chromosomes in yeast cells). These vectors also contain suitable restriction sites to clone foreign DNA and genes to be used as selectable markers.
Human artificial chromosome
Human artificial chromosomes may be potentially useful as gene transfer vectors for gene delivery into human cells and a tool for expression studies and determining human chromosome function. It can carry very large DNA fragments (there is no upper limit on size for practical purposes). Therefore, it does not have the problem of limited cloning capacity of other vectors, and it also avoids possible insertional mutagenesis caused by integration into host chromosomes by a viral vector.
Animal and plant viral vectors that infect plant and animal cells have also been manipulated to introduce foreign genes into plant and animal cells. The natural ability of viruses to bind to cells, introduce their DNA, and replicate has made them ideal vehicles for transferring foreign DNA into eukaryotic cells in culture. A vector based on Simian virus 40 (SV40) was used in the first cloning experiment involving mammals. Several vectors based on viruses like Adenoviruses and Papillomavirus have been used to clone genes in mammals. At present, retroviral vectors are popular for cloning genes in mammalian cells. In the case of plant transformation, viruses, including the Cauliflower Mosaic Virus, Tobacco Mosaic Virus, and Gemini Viruses, have been used with some success.
Summary of DNA Cloning
Figure \(\PageIndex{7}\) summarizes the basic cloning methods most widely used in biochemistry laboratories. Foreign DNA is isolated or amplified using PCR to obtain enough material for cloning. The DNA is purified and cut with restriction enzymes and then mixed with a vector cut with the same restriction enzymes. The DNA can then be stitched back together with DNA ligase. The DNA can then be transformed into a host system, often bacteria, to grow large quantities of the plasmid containing the cloned DNA.
Restriction fragment patterning and DNA sequencing can be used to validate the cloned material. For a Video Tutorial on DNA Cloning visit HHMI - BioInteractive.

Plasmids with foreign DNA inserted into them are called recombinant DNA molecules because they contain new combinations of genetic material. Proteins that are produced from recombinant DNA molecules are called recombinant proteins. Not all recombinant plasmids are capable of expressing genes. Plasmids may also be engineered to express proteins only when stimulated by certain environmental factors so that scientists can control the expression of the recombinant proteins.
Reproductive Cloning
Reproductive cloning is a method used to make a clone or an identical copy of an entire multicellular organism. Most multicellular organisms undergo reproduction by sexual means, which involves the contribution of DNA from two individuals (parents), making it impossible to generate an identical copy or a clone of either parent. Recent advances in biotechnology have made it possible to clone mammals reproductively in the laboratory.
Natural sexual reproduction involves the union, during fertilization, of a sperm and an egg. Each of these gametes is haploid, meaning they contain one set of chromosomes in their nuclei. The resulting cell, or zygote, is then diploid and contains two sets of chromosomes. This cell divides mitotically to produce a multicellular organism. However, the union of just any two cells cannot produce a viable zygote; there are components in the cytoplasm of the egg cell that are essential for the early development of the embryo during its first few cell divisions. Without these provisions, there would be no subsequent development. Therefore, a diploid genetic complement and an egg cytoplasm are required to produce a new individual. The approach to producing an artificially cloned individual is to take the egg cell of one individual and remove the haploid nucleus. Then, a diploid nucleus from the body cell of a second individual, the donor, is put into the egg cell. The egg is then stimulated to divide so that development proceeds. This sounds simple, but it takes many attempts to complete each step successfully.
The first cloned agricultural animal was Dolly, a sheep who was born in 1996 (see Figure \(\PageIndex{8}\) below). The success rate of reproductive cloning at the time was very low. Dolly lived for six years and died of a lung tumor. There was speculation that because the cell DNA that gave rise to Dolly came from an older individual, the age of the DNA may have affected her life expectancy. Since Dolly, several species of animals (such as horses, bulls, and goats) have been successfully cloned.
There have been attempts at producing cloned human embryos as sources of embryonic stem cells. In the procedure, the DNA from an adult human is introduced into a human egg cell, which is then stimulated to divide. The technology is similar to the technology used to produce Dolly, but the embryo is never implanted into a surrogate mother. The cells produced are called embryonic stem cells because they can develop into many different kinds of cells, such as muscle or nerve cells. The stem cells could be used for research and provide therapeutic applications, such as replacing damaged tissues. The benefit of cloning in this instance is that the cells used to regenerate new tissues would be a perfect match to the donor of the original DNA. For example, a leukemia patient would not require a sibling with a tissue match for a bone marrow transplant.

To create Dolly, the nucleus was removed from a donor egg cell. The enucleated egg was placed next to the other cell, and they were shocked to fuse. They were shocked again to start the division. The cells were allowed to divide for several days until an early embryonic stage was reached before being implanted in a surrogate mother.
Why was Dolly a Finn-Dorset and not a Scottish Blackface sheep?
Because even though the original cell came from a Scottish Blackface sheep and the surrogate mother was a Scottish Blackface, the DNA came from a Finn-Dorset.
Genetic Engineering
Genetic engineering uses recombinant DNA technology to modify an organism’s DNA to achieve desirable traits. Adding foreign DNA in the form of recombinant DNA vectors generated by molecular cloning is the most common method of genetic engineering. An organism that receives the recombinant DNA is called a genetically modified organism (GMO). The host organism is called transgenic if the foreign DNA that is introduced comes from a different species. Bacteria, plants, and animals have been genetically modified since the early 1970s for academic, medical, agricultural, and industrial purposes.
Watch this short video explaining how scientists create a transgenic animal.
Although the classic methods of studying the function of genes began with a given phenotype and determined the genetic basis of that phenotype, modern techniques allow researchers to start at the DNA sequence level and ask: “What does this gene or DNA element do?” This technique, called reverse genetics, reverses the classical genetic methodology. One example of this method is analogous to damaging a body part to determine its function. An insect that loses a wing cannot fly, meaning the wing’s function is flight. The classic genetic method compares insects that cannot fly with insects that can fly and observes that the non-flying insects have lost wings. Similarly, in a reverse genetics approach, mutating or deleting genes provides researchers clues about gene function. Alternately, reverse genetics can be used to cause a gene to overexpress itself to determine what phenotypic effects may occur.
CRISPR Technology
CRISPR stands for clustered regularly interspaced short palindromic repeats and represents a family of DNA sequences found within the genomes of prokaryotic organisms such as bacteria and archaea. These sequences are derived from DNA fragments of bacteriophages that have previously infected the prokaryote and are used to detect and destroy DNA from similar phages during subsequent infections. Hence, these sequences play a key role in the prokaryotes' antiviral defense system. Figure \(\PageIndex{9}\) shows the crystal structure of a CRISPR RNA-guided surveillance complex, Cascade, bound to a ssDNA target,
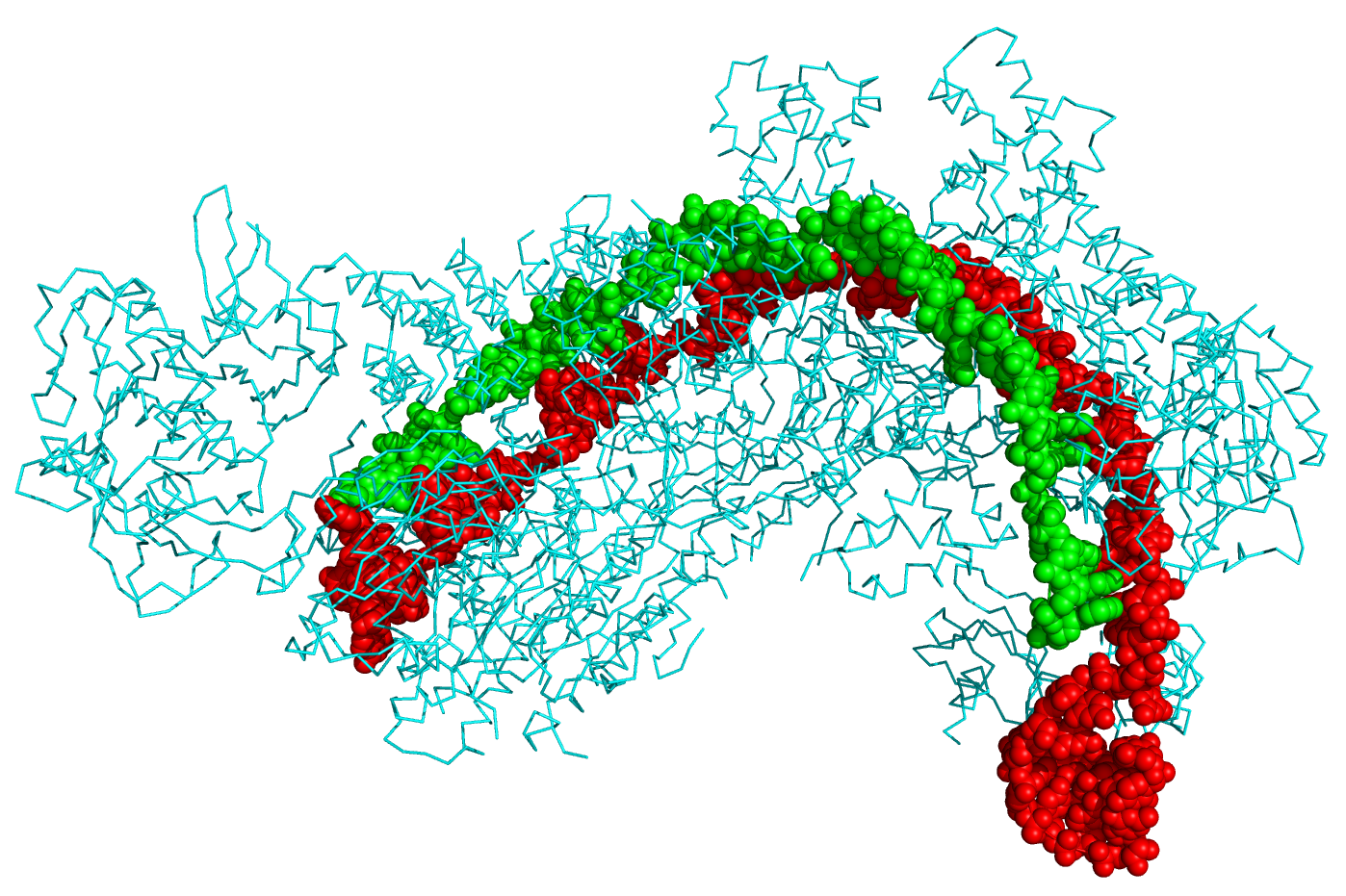
Figure \(\PageIndex{9}\): Crystal structure of a CRISPR RNA-guided surveillance complex, Cascade, bound to a ssDNA target. CRISPR system Cascade protein subunits CasA, CasB, CasC, CasD, and CasE (cyan) bound to CRISPR RNA (green) and viral DNA (red) based on PDB 4QYZ and rendered with PyMOL. Image from Boghog
Cas9 (or "CRISPR-associated protein 9") is an enzyme that uses CRISPR sequences as a guide to recognize and cleave specific strands of DNA that are complementary to the CRISPR sequence. Cas9 enzymes and CRISPR sequences form the basis of a technology known as CRISPR-Cas9 that can be used to edit genes within organisms. This editing process has a wide variety of applications, including basic biological research, developing biotechnology products, and treating diseases. Figure \(\PageIndex{10}\)s shows a diagram of the CRISPR prokaryotic antiviral defense mechanism
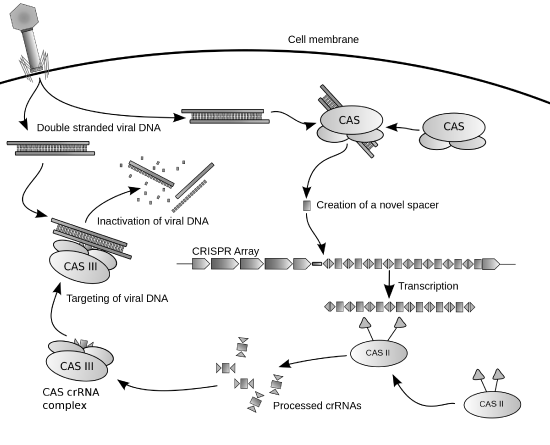
(\PageIndex{10}\): Diagram of the CRISPR prokaryotic antiviral defense mechanism. Image by James Atmos
The CRISPR-Cas system is a prokaryotic immune system that confers resistance to foreign genetic elements, such as those present within plasmids and phages that provide acquired immunity. RNA harboring the spacer sequence helps Cas (CRISPR-associated) proteins recognize and cut foreign pathogenic DNA. Other RNA-guided Cas proteins cut foreign RNA. CRISPR is found in approximately 50% of sequenced bacterial genomes and nearly 90% of sequenced archaea.
Summary
This chapter provides an integrated overview of the strategies and tools used to isolate, manipulate, and express DNA for research and biotechnology applications. It begins by differentiating molecular cloning—the process of copying short segments of DNA—and reproductive cloning, which involves creating a whole organism that is genetically identical to the donor. The focus is on molecular cloning techniques that enable researchers to generate multiple copies of specific genes for analysis and expression in various host cells.
Central to molecular cloning is the use of cloning vectors—small, stable DNA molecules (typically plasmids, bacteriophages, cosmids, bacterial artificial chromosomes, yeast artificial chromosomes, or even human artificial chromosomes) engineered to carry foreign DNA fragments. These vectors incorporate essential features such as origins of replication, selectable markers (e.g., antibiotic resistance genes), and multiple cloning sites (MCSs) that facilitate the insertion and subsequent manipulation of target genes. Depending on experimental needs, vectors may also include reporter genes or regulatory elements to control and monitor gene expression.
The chapter explains how restriction enzymes (restriction endonucleases) are used as molecular scissors to cut DNA at specific palindromic sequences, generating fragments with either sticky or blunt ends. These fragments can be ligated into vector DNA, forming recombinant molecules that are then introduced into host cells—most commonly Escherichia coli—for propagation and analysis. Alternative cloning methods, such as TOPO cloning and Gateway recombination, are discussed as streamlined approaches that bypass traditional restriction digestion and ligation steps.
In addition to molecular cloning, the chapter covers reproductive cloning, highlighting the process of somatic cell nuclear transfer used to create clones such as Dolly the sheep. This section contrasts the technical challenges and biological implications of cloning entire organisms versus cloning specific genes.
The discussion then shifts to genetic engineering, where recombinant DNA technology is employed to create genetically modified (GM) and transgenic organisms. This reverse genetics approach allows researchers to study gene function by introducing, deleting, or modifying genes to observe resulting phenotypic changes.
A significant portion of the chapter is devoted to the CRISPR-Cas system—a natural adaptive immune mechanism found in prokaryotes that has been repurposed as a powerful tool for genome editing. The CRISPR-Cas9 complex uses a guide RNA to direct the Cas9 nuclease to specific DNA sequences, enabling targeted double-strand breaks that can be exploited for gene knockout, correction, or insertion. This technology has revolutionized the field by providing unprecedented precision and ease in modifying genetic information, with broad applications in research, medicine, and agriculture.
Overall, the chapter ties together the fundamental principles of DNA manipulation—from isolation and cloning to precise gene editing—underscoring how these techniques have transformed our ability to read, write, and edit the genetic code.