
4.10: Protein Aggregates - Amyloids, Prions and Intracellular Granules
- Page ID
- 71657


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Differentiate Normal vs. Aberrant Aggregation:
- Explain the difference between protein multimerization (e.g., dimers, trimers, filaments) in the native state and the formation of pathological aggregates such as amyloid fibrils.
- Recognize the role of conformational flexibility and alternative folding in driving the formation of aggregates.
-
Describe the Mechanism of Amyloid Fibril Formation:
- Illustrate how proteins that can adopt two stable or metastable conformations convert from a native (often α-helical) form to an abnormal (β-sheet-rich) conformation.
- Explain the nucleation process and subsequent fibril elongation via templated conformational conversion.
-
Analyze the Role of Molecular Chaperones:
- Discuss how molecular chaperones assist in proper protein folding and prevent aggregation.
- Understand how chaperone malfunction or overload can contribute to the accumulation of misfolded proteins.
-
Link Protein Aggregation to Disease Pathogenesis:
- Identify the connection between amyloid aggregates and neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s, familial amyloidotic polyneuropathy, prion diseases).
- Discuss how mutations, environmental factors, or post-translational modifications can shift the balance toward aggregation and disease.
-
Interpret Structural and Experimental Data:
- Utilize structural models (e.g., cryo-EM and NMR data) to understand the common β-sheet architecture of amyloid fibrils.
- Evaluate experimental approaches (e.g., in vitro folding/refolding experiments and mutagenesis studies) used to investigate the aggregation process.
-
Examine the Specificity and Regulation of Aggregation:
- Explain how specific amino acid sequences and even single-point mutations can program a protein to aggregate.
- Analyze factors that control the transition from soluble protein states to reversible phase-separated granules or irreversible fibrils.
-
Understand Phase Separation and Its Biological Implications:
- Describe how liquid–liquid phase separation leads to the formation of intracellular granules and condensates.
- Compare these reversible assemblies with the irreversible aggregation seen in amyloid-related diseases.
-
Evaluate Therapeutic Implications:
- Discuss potential therapeutic strategies aimed at modulating protein aggregation or stabilizing the native state.
- Consider how insights from aggregate formation can lead to the development of diagnostics and treatments for neurodegenerative disorders.
These learning goals encourage a deep understanding of the thermodynamic, structural, and cellular factors that influence protein aggregation, linking molecular-level phenomena to clinical outcomes and potential therapeutic approaches.
Introduction to Protein Aggregates
We have studied different types of protein aggregation, including aggregation of the native state (to form dimers, trimers, multimers, and filaments). We've also studied how misfolded proteins can aggregate and how a whole family of molecular chaperones helps newly synthesized and misfolded proteins fold correctly. But what happens if a protein can fold two reasonably stable or metastable structures of starkly different conformation? A class of proteins known as prions or amyloid proteins exhibits this characteristic. Their "alternative" conformation can bind to the "normal" structure, causing it to flip to the alternative conformation. This can then seed the continuation of the process, which ultimately forms large aggregates with a fibrillar structure. This process leads to aberrant cell function and, when it occurs in neurons, can lead to a variety of brain diseases such as Alzheimer's Disease.
Protein aggregates complicate the work of researchers studying protein folding in vitro and attempting to express human proteins in prokaryotes, such as E. coli, in vivo. This often results in the formation of large protein aggregates called inclusion bodies. Instead of viewing these aggregates as unwanted "junk", some study them avidly. It turns out that these aggregates are not as non-specific as was earlier believed. In addition, understanding how and when they form will give us clues into the etiology and treatment of some of the most debilitating and feared diseases.
Specificity of Aggregate Formation
In the early 1970s, it was demonstrated that chymotrypsinogen could not be folded in vitro without forming aggregates. An intermediate was presumed to have formed that, if present in high concentration, would aggregate irreversibly instead of folding to the native state. Refolding of tryptophanase showed that it aggregated only with itself, suggesting specificity. In the 1980s, a single amino acid folding mutant was found in a viral protein. Both the normal and mutant viral proteins unfolded at high temperatures. Still, only the mutant would aggregate at high temperatures, suggesting that aggregation could be programmed into or out of a gene. Also, a single amino acid change in bovine growth hormone prevented aggregation without affecting correct folding.
This knowledge of protein folding and aggregation was soon applied to understanding several diseases in which protein aggregates were observed, either initiating or being associated with the diseases. These protein aggregates were termed "amyloid deposits" and appeared to be associated with, and perhaps causative of, several neurodegenerative diseases. The term amyloid was first used by the German pathologist Rudolf Virchow. In 1853, he described waxy tissue deposits associated with eosinophils (a type of immune cell). These deposits resembled starch (made of amylose and amylopectin), so he termed them amyloid. However, all known amyloid deposits are composed of protein, not starch.
It now appears that these diseases are likely caused by improper protein folding and subsequent aggregation. Except in rare inherited diseases, amyloid deposits are composed of normal, wild-type proteins (not mutants) that undergo conformational changes to form monomers. These monomers then catalyze the conversion of normal monomers into the altered form, which polymerize into fibrils. Sometimes, in inherited conditions or when mutations occur in a specific protein, amyloid deposits consist of the mutant protein. The proteins in these deposited fibers are composed predominantly of β sheets perpendicular to the fiber axis. Sometimes, the protein's monomeric "normal conformation" has little beta sheet structure.
Figure \(\PageIndex{1}\) shows a simplified model of how a normal protein with a "normal" conformation, enriched in this case for illustration in alpha-helices, can form fibrils of abnormal monomers, which are highly enriched in beta sheets. These can self-associate to form large insoluble protein "amyloid" fibers. Note the green arrows representing beta-strands.

In the misfolded state, proteins have an increased propensity to oligomerize through interactions of their metastable beta-sheet domains. These can form more stable beta-sheet states, and the ensuing oligomers act as nuclei for subsequent elongation reactions, forming so-called protofibrils. The final amyloid fibril usually consists of many intertwined protofibrils.
Diseases of Protein Aggregates
We'll now describe a series of diseases caused by or highly associated with fibril formation from normal soluble proteins. For each one, we will present the best available structure of the amyloid fiber obtained mostly through cryo-EM. What's amazing is that, at first glance, all of the amyloid fiber structures have an astonishingly similar structural appearance. We present them not to be redundant but to illustrate how natural processes can yield a common structural outcome, often lethal, from a great diversity of protein structures. The amyloid fibers are so structurally similar that a common therapy to prevent their formation may be developed.
Familial amyloidotic polyneuropathy (FAP)
This affects 1/10,00 to 1/100,000 people. The monomer protein involved is transthyretin (147 amino acids, MW 15,887), which normally exists in the blood as a homotetramer (a dimer of dimers). Figure \(\PageIndex{2}\) shows the structure of the dimer (6fxu). Note that each monomer contains mostly beta structure. The protein binds L-thyroxine, and around 40% of blood plasma transthyretin is bound to retinol-binding protein.

In mildly acidic conditions in vitro, the equilibrium between the tetramer and monomer is shifted toward the monomer, which can aggregate into fibrils. Monomer aggregation could be promoted by a possible transition to a molten globule-like state (as discussed previously for lactalbumin). This secondary but loosely packed tertiary structure has more exposed hydrophobic groups. If the concentration is high enough, the molten globules aggregate. In individuals with the disease, mutations in the protein destabilize the tetramer, shifting the equilibrium toward the monomer, which presumably increases molten globule formation and aggregation. Specifically, Val30Met and Leu55Pro mutations promote the dissociation of the tetramer and the formation of aggregates. Conversely, Thr119Met inhibits tetramer dissociation—amyloid aggregates deposit in the heart, lungs, kidneys, and other organs, leading to death. Figure \(\PageIndex{3}\) shows how the transthyretin dimer could be stabilized to form monomers or dimers, leading to fibril formation.

Figure \(\PageIndex{4}\) shows the intramolecular hydrogen bonds bonds within monomers (-----) and intermolecular hydrogen bonds between monomers (-----) in the transthyretin dimer. We present this figure to remind readers that in all of the complex amyloid fibrillar structures presented below, the bulk of hydrogen bonds are "intermolecular" between adjacent monomers in their extended states (phi/psi angles consistent with beta-structure) within the fibrillar structure.


Figure \(\PageIndex{5}\) shows an interactive iCn3D model of the Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis (6SDZ). Each separate monomer is shown in a different color. The static image below shows arrows indicating beta strands forming hydrogen bonds from the main chain amide Hs and carbonyl Os to another main chain atom on an adjacent monomer.

Note the beautiful but, unfortunately, deadly array of adjacent extended monomeric chains (each colored differently) that form hydrogen bonds with adjacent extended monomers to stabilize the fibrillar structure.
Light Chain Amyloidosis - AL amyloidosis (amyloidosis from the light chain)
The light chain (MW approx 25,000) is a normal component of circulating immunoglobulin antibody (protein) molecules. Each contains two light and two heavy chains. We will discuss the structure of antibodies in detail in Chapter 5.5. Needless to say, antibodies are very diverse molecules. The immune system can generate antibodies to recognize almost any foreign molecule. The light chains of antibodies are incredibly diverse and variable, despite all having two immunoglobulin domains. Each domain consists of approximately 110 amino acids, organized into two layers of β-sheets. Each layer contains 3-5 antiparallel β-strands, with a disulfide bond connecting the two layers. As a result, they exhibit significant β-structure in their monomeric form. The large diversity in light chains is partly generated by recombining gene fragments to produce individual light chains. Some variants of the light chains (λ1, λ2, λ3, λ6, and κ1) are associated with AL amyloidosis, a potentially fatal disease. Mutants in the light chain can cause a destabilization of the native state to a state similar to a molten globule, which then conformationally converts to a structure that aggregates into amyloid fibers. These can be deposited in various tissues.
As shown above, the pathway that a simple 25K monomer takes to produce such a complex but regular structure must start with a simple dimer formation between two monomers. Figure \(\PageIndex{6}\) shows an interactive iCn3D model of the normal conformation of a λ6a light chain dimer (6mg4). The λ6a light chain variant is more prone to aggregate to form fibrils.
.png?revision=1&size=bestfit&width=380&height=240)
 Figure \(\PageIndex{6}\): Full-length human lambda-6A light chain dimer showing IgG fold domains (6mg4). (Copyright; author via source).
Figure \(\PageIndex{6}\): Full-length human lambda-6A light chain dimer showing IgG fold domains (6mg4). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...MNsiddm4RAdKn8
One light chain in the dimer is shown in blue, with one IgG fold domain in light blue and the other in dark blue. The other light in the dimer is shown in shades of magenta to show the two domains. Normally, light chains don't form dimers; instead, they associate with heavy chains to form full IgG antibodies. Free light chains will form dimers if not associated with a heavy chain, altering conformation and forming amyloid fibrils.
Figure \(\PageIndex{7}\) shows an interactive iCn3D model of the AL amyloid fibril from a lambda 3 light chain in conformation A (6Z1O). Each separate monomer is represented in a distinct color.

Figure \(\PageIndex{7}\): AL amyloid fibril from a lambda 3 light chain in conformation A (6Z1O). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...pSu2Nm714gvjh7
Figure \(\PageIndex{8}\) shows a comparison of the secondary structures of the native conformation of the light chain and those found in the two different fibrillar forms (A and B) of the amyloid fibrils (panel a). Remember that each light chain has two IgG domains, each having two sets of β-sheets containing 3-5 antiparallel β-strands.

Panel b shows the alignment of six fibrillar light chain proteins in conformation A. Panel C shows the backbones' trace and the side chains' local environment in a single light chain from the fibrillar A conformations. It shows cross-β-sheets interactions with parallel hydrogen bonds across strands. Note the disulfide that connects Cys 22 and Cys 87 on adjacent strands. The circles show surface side chains. Note that they are enriched in green (polar) and blue/red (basic/acidic) regions, with some hydrophobic patches (e.g., V10-L14). Some are pairs, such as D24 and R28. Panel D shows the electrostatic surface of a single light chain from the fibrillar A conformation, with red indicating negative and blue positive.
Alzheimer's Disease (AD)
This disease accounts for 60-80% of dementia cases. Brains in Alzheimer's patients have amyloid deposits of the amyloid beta (Aβ) protein and aggregates of a protein called tau. The origin of this disease remains unresolved. Some believe aggregates of the amyloid beta protein cause the disease. Others suspect that tau plays a role in forming tau bundles. Still others suggest an infectious disease agent (more on that later). Regardless of the underlying cause, the amyloid beta protein aggregates are neurotoxic and, at the very least, correlate with, if not cause, the disease.
The amyloid aggregates in Alzheimer's disease begin with a change in a monomeric protein normally found in neuronal membranes. The protein, called β-amyloid precursor protein (BAPP or simply APP), is a transmembrane protein. A slightly truncated, soluble form is secreted from cells and found in the extracellular fluid (cerebrospinal fluid and blood). The normal function of these APP proteins is not yet clear. An endoprotease cleaves a small 40-42 amino acid fragment from this protein, named the amyloid beta (Aβ) protein. Figure \(\PageIndex{9}\) shows the normal processing of the amyloid precursor protein APP (left) and the abnormal, amyloidogenic form (right).
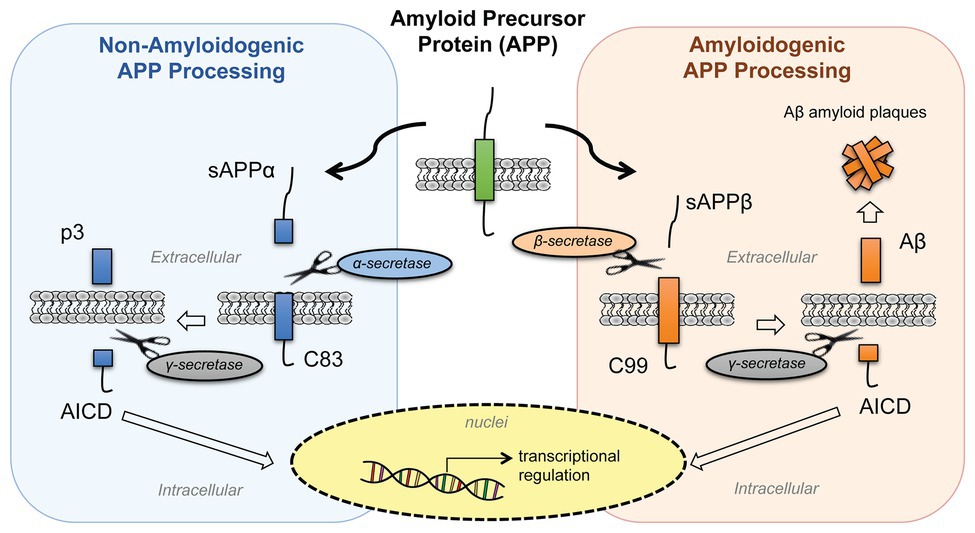
In a "normal" processing pathway, the proteases α- and γ-secretase release two variant peptides into the extracellular environment: soluble amyloid precursor protein cleaved by α-secretase (sAPPα) and p3 fragments. In the amyloidogenic processing pathway, β- and γ-secretases release soluble amyloid precursor protein cleaved by β-secretase (sAPPβ) and β-amyloid (Aβ) peptides. Both pathways release the same intracellular domain, AICD, which moves to the nucleus and acts as a transcription factor to regulate gene expression. The β-amyloid (Aβ) peptides aggregate to form the fibrillar plaque.
The amyloid beta (Aβ) protein, or a mutant form of it, aggregates to form beta-sheet-containing fibrils in Alzheimer's disease. The NMR solution structure of the monomer amyloid beta-peptide (1-42) is shown in Figure \(\PageIndex{10}\). Note the absence of any beta structure.

Several mutations in different proteins have been linked to Alzheimer's disease, but they all appear to increase the production or deposition of the amyloid beta protein. These deposited plaques are extracellular and have been shown to cause neuronal damage. They are located in brain regions essential for memory and cognition. The APP gene is located on chromosome 21, the same chromosome that is present in an extra copy (trisomy 21) in Down syndrome, which is characterized by symptoms including presenile dementia and amyloid plaques. Aggregate formation appears to be driven by increased APP expression and, hence, amyloid beta protein. Additionally, some mutants may serve to destabilize the amyloid beta protein, thereby increasing its aggregation.
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of the prevalent amyloid-beta fibril structure from Alzheimer's disease brain tissue (6W0O).

Figure \(\PageIndex{11}\): Amyloid-beta(1-40) fibril derived from Alzheimer's disease cortical tissue (6W0O). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...NZCX9sAhnFPiB6
The tau protein (758 amino acids, MW 78,928), much larger than the other proteins we discuss in this chapter, has also been implicated as a cause or factor in Alzheimer's Disease. It facilitates microtubule assembly and stability, as well as other neuronal functions (see Chapter 5). Since its C-terminus binds microtubules in axons and the N-terminus binds the plasma membrane, it might link both. The cytoskeleton of neurons is disrupted in Alzheimer's and in patients with other neurodegenerative diseases. When tau tangles are present, these diseases are also referred to as tauopathies. One tauopathy is chronic traumatic encephalopathy (CTE) caused by repetitive head impacts (from contact sports or physical abuse) or concussions arising from explosions in combat. No full-length structure of tau has been determined to date. The structures of a predicted model of solution phase monomeric tau and tau fibers from the brain of patients with neurodegenerative diseases (Alzheimer's, CTE, and Corticobasal Degeneration - CBD) are shown in Figure \(\PageIndex{12}\).

The predicted structure of the monomeric protein (using AlphaFold) is almost completely devoid of secondary structure. The tau structures in other tauopathies are similar but distinct. In CBD tau fibers, there are four microtubule-binding repeats (4R). Pick's Disease, another tauopathy, has three repeats (3R), while taus in AD and CTE are 3R or 4R.
The fibril cores of tau in both CBD (Lys 274-Glu380, which contains the end of R1 and R2-R4) and Alzheimer's Disease (AD) contain around 13% glycine residue. These allow the main chain flexibility and intersheet packing, enabling the large conformational changes necessary for beta structure formation and fibril formation. Repeats of the PGGG motif allow sharp turns or extended chains. Valine (around 10%) and isoleucine, leucine, and phenylalanine facilitate inter-sheet packing through induced dipole-induced dipole interactions and the hydrophobic effect. Certain tau fibrils in CBD contain hydrophilic pockets that bind molecules that have yet to be elucidated and that might seed the nucleation of fibrils. The cavity has three lysines and a histidine, so any ligands that bind are likely anionic in nature. In addition, recent evidence suggests that different combinations of post-translational modifications, including ubiquitinylation and acetylation of lysines 311, 317, 321, 343, and 353, may lead to distinct tau fibril structures. CTE tau protein in tangles appears to be hyperphosphorylated.
Figure \(\PageIndex{13}\) shows an interactive iCn3D model of a paired helical tau filament from Alzheimer's Disease human brain tissue (6VHL).
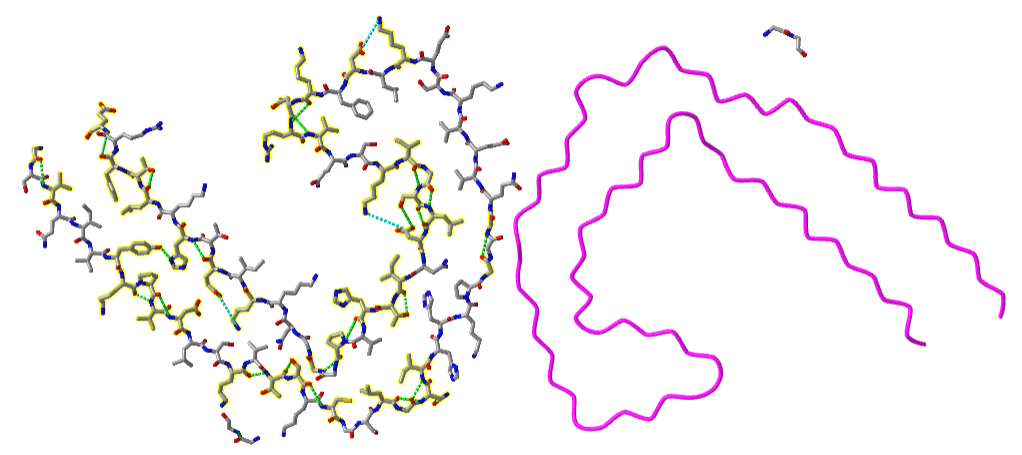
Figure \(\PageIndex{13}\): Paired helical filament of Tau (6VHL) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...NHsZdhDJNentg9
Zoom into the interactive images to see the H bonds within one of the filaments. Note that the left filament's hydrogen bonds (green dotted lines) are between side chains and not between backbone amide Hs and carbonyl Os. These are pointing above and below the backbone plane, where they could interact with other chains above and below to form the multi-chain fibers seen in the examples above.
Figure \(\PageIndex{14}\) shows an interactive iCn3D model of a singlet Tau fibril obtained from corticobasal degenerated human brain tissue (6VHA).
.png?revision=1)
Figure \(\PageIndex{14}\): Singlet Tau fibril from corticobasal degeneration of human brain tissue (6VHA). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...Bbxr2Z1caGqnG8
The backbone of the C-terminal amino acid (367-380) of each of the three separate chains of the fibril is shown in CPK colors, and hydrogen bonds between the strands that form the parallel beta strands are shown as green dashes.
Progress has been incredibly slow on ways to treat Alzheimer's. Most methods focus on reducing amyloid beta production and aggregation by identifying small molecules that inhibit specific steps in its production, including secretase cleavage and the subsequent conformational changes necessary to form amyloid fibers. But what if the amyloid aggregates are secondary to the primary cause? What if the amyloid beta protein overproduction was the brain's response to defend against the cause?
Alzheimer's patients have aggregates of the amyloid beta and tau proteins. Do these aggregates cause the disease? Maybe. Maybe not! It's very easy to confuse correlation with causation. Also, it isn't easy to go against present beliefs. If you seek funding for an alternative cause and your funding derives from grant applications reviewed by experts in the field who are firmly in the amyloid beta and tau "camps", you are not likely to get funding.
Here is an interesting story that illustrates a source of bias we all face, including AD researchers. During World War II, when British Royal Air Force (RAF) planes returned from bombing missions, they were inspected for damage caused by German anti-aircraft fire. To protect pilots from harm, the planes were reinforced in the areas that had received incoming enemy fire. It made sense and was intuitive until a statistician, Abraham Wald, realized that the damaged planes had returned safely. Instead of looking at the sites hit by enemy fire, he recommended looking at the sites NOT hit. In those areas, it was likely the hits were fatal. The fatal sites were not included in the collected sample data, so the sample was biased. Selectively attending to certain data or ideas while ignoring other information is called selection bias. We all face this, and scientists studying and funding AD also do.
Another problem that has emerged in AD research is scientific fraud. Decades of research have yielded no treatments that significantly impact the disease. One reason, in addition to selection bias, is scientific fraud. Charles Piller has written about extensive fraud using falsified data collected by key and leading researchers in the field. This has had an enormous and deleterious effect not only on AD research but on the victims of this disease as well.
Lewy Body and Parkinson's Disease
α-Synuclein (140 amino acids, MW 14,460) is expressed in the brain and presynaptic terminals in the central nervous system, but also in more distal neurons, and is involved in the regulation of neurotransmitter release and in the synaptic vesicles that hold them. Its aggregation is a cause or consequence of Parkinson's Disease and Lewy Body Dementia. It's found in the cytoplasm and the nucleus and is also secreted. Figure \(\PageIndex{15}\) shows an NMR solution structure (left) and AlphaFold-predicted structure (right) for this protein, which in solutions is so disordered that no full crystal structure has been determined.

Figure \(\PageIndex{16}\) shows an interactive iCn3D model of an amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy (6A6B). Two protofilaments with clearly Greek key topologies are shown.

Figure \(\PageIndex{16}\): Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy (6A6B). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...SbQq7fhFpQ9K4A
Click below for more information!
- Answer
-
A substantial body of evidence suggests that Parkinson's Disease may have its origin in the gut or even the nose. Changes in the gut lining, caused by and/or leading to inflammation, as well as an altered gut microbiome (which can also cause inflammation), may lead to misfolding of gut alpha-synuclein. Sources of gut damage and inflammation include an altered gut microbiome, Helicobacter pylori infection, and the use of nonsteroidal anti-inflammatory drugs (NSAIDs). Altered synuclein formed in the gut could migrate through the vagal nerve from the gut to the brain. The processes involved are outlined in Figure \(\PageIndex{17a}\) below.

Figure \(\PageIndex{17a}\): Model of gut-originating, inflammation-driven PD pathogenesis. Houser, M.C., Tansey, M.G. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis?. npj Parkinson's Disease 3, 3 (2017). https://doi.org/10.1038/s41531-016-0002-0. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/
In a susceptible individual, inflammatory triggers (1) initiate immune responses in the gut that deleteriously impact the microbiota, increase intestinal permeability, and induce increased expression and aggregation of αSYN (2). Synucleinopathy may be transmitted from the gut to the brain via the vagus nerve (3b), and chronic intestinal inflammation and permeability can promote systemic inflammation, which, in turn, can increase blood-brain barrier permeability (3a). Intestinal inflammation, systemic inflammation, and synuclein pathology in the brain promote neuroinflammation (4), which drives the neurodegeneration that characterizes PD (5).
The vagal nerve is a very long nerve that consists of a bundle of sensory (80%) and motor (20%) neurons. It controls autonomic processes, such as breathing, blood pressure, heart rate, gut motility, and neural responses related to feeding and drinking. Sensory neurons of the vagal nerve are found in the digestive, cardiovascular, and pulmonary systems. Figure \(\PageIndex{17b}\) shows the vagal nerve and its branches.

Figure \(\PageIndex{17b}\): The Vagal Nerve. R. Douglas Fields. How Our Longest Nerve Orchestrates the Mind-Body Connection. QuantaMagazine. August 26, 2024. https://www.quantamagazine.org/how-o...tion-20240826/. With Permission from Quanta Magazine.
Another recent study suggests that altered alpha-synuclein might arise in the nose from the protein's interaction with environmental toxins. These include trichloroethylene (TCE) and perchloroethylene (PCE), used in dry cleaning and degreasing, paraquat (an herbicide), and air pollution.
Transmissible spongiform encephalopathies (TSEs) - Prion Diseases
Prion diseases are another set of brain diseases resulting from aggregates of monomers of the prion protein (PrPc). As with the examples above, the aggregates form amyloid beta fibrils. The prion diseases include scrapie in sheep, bovine spongiform encephalopathy (mad cow disease), and in humans, Creutzfeldt-Jakob Disease (CJD), Fatal Familial Insomnia (FFI), Gerstmann-Straussler-Scheinker Syndrome, and Kuru (associated with cannibalism). In these fatal diseases, the brain, on autopsy, resembles a sponge with holes (hence the name spongiform). In contrast to the diseases described above, these can be transmitted from one animal to another, but typically not between species. (However, consider the controversy surrounding mad cow disease.) Also, the infectious agent can self-replicate in vivo. The logical conclusion is that a virus (slow-acting) is the causative agent. However, the infectious agent survives radiation, heat, chemical agents, and enzymes designed to kill viruses and their associated nucleic acids. Mathematical analyses suggested that the infectious agent in such diseases could be nothing more than a protein. Stanley B. Prusiner, in the '80s, isolated just such a protein, which he named a prion, for a proteinaceous infectious agent. In October 1997, he was awarded the Nobel Prize in Medicine.
The normal monomeric prion protein, PrPc (253 amino acids, MW 27,661), is highly conserved in mammals and is widely expressed in embryogenesis. Expression is highest in the central nervous system. The protein's normal function remains unclear. It is a physiological substrate to a particular membrane receptor (the Gpr 126 G protein-coupled receptor). Knocking out the gene shows that the normal protein is involved in synapse structure/function, myelination of neurons, and circadian rhythms, probably by acting as a transcription factor. It also helps regulate Cu2+ and Zn2+ levels in the central nervous system. The protein is cleaved, and a 209-amino-acid fragment is bound to the extracellular side of the neuronal membrane, anchored by the attachment of a lipid (GPI) anchor.
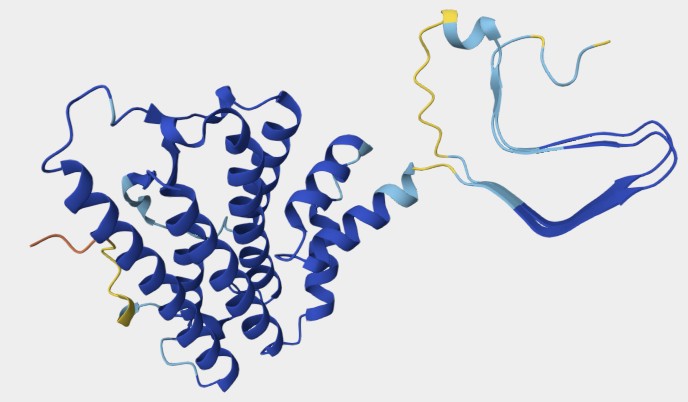
The N-terminal residues (23-124) are flexible, followed by residues 125-231, which are mostly alpha-helical. There is a disulfide between Cys179 and Cys214. The PrPc (without the PI link) is water soluble, a monomer, protease-sensitive, and consists of around 45% alpha helix and 3% beta sheet. Given its highly disordered structure, no full-length crystal structure of the protein has been determined to date. The solution NMR structure of residues 125-231 has been determined, and the structure of the full protein has been modeled with AlphaFold. These structures are shown in Figure \(\PageIndex{18}\).

The blue helices and gold loops in the computer model consist of the same amino acids (125-231) as the NMR solution model on the left-hand side of Figure \(\PageIndex{2}\).
The problem in transmissible spongiform encephalopathies (TSEs) is that neurotoxic amyloid-like protein aggregates form. The protein found in plaques (in cases other than those inherited) has the same primary sequence as PrPc but a different secondary and, presumably, tertiary structure. The protein found in the plaques, called PrPsc (the scrapie form of the normal protein), is insoluble in aqueous solutions, protease-resistant, and has a high beta-sheet content (43%) and a lower alpha-helix content (30%) than the normal version of the protein, PrPc.
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of the cryo-EM structure of an amyloid fibril formed by full-length human prion protein (6LNI). Each line represents a PrPsc chain from amino acids 170-229, which is the core of the fibril.

Figure \(\PageIndex{19}\): Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. (6LNI) (Copyright; author via source).
Click the image for a popup or use this external link https://structure.ncbi.nlm.nih.gov/i...ByT4rL33brHRY7
The aligned zig-zag lines indicate beta strands that are hydrogen-bonded to the adjacent strands. The two alpha helices in the C-terminal domain become beta strands.
A genetic, inheritable form of the disease also exists, in which a mutant form of the PrPc occurs, whose normal structure is destabilized by the mutation. The aggregates caused by the mutant form of the disease are understandable in light of the other diseases we discussed above. How does the normal PrPc form PrPsc? Evidence shows that if radiolabeled PrP*c from scrapie-free cells is added to unlabeled PrPsc from scrapie-infected cells, the PrP*c is converted to PrP*sc! It appears that the PrPc protein has two forms, not much different in energy, one composed mostly of alpha helices and the other of beta sheets. A dimer of PrPc.PrPsc might form, destabilizing PrP*c, causing and driving a conformational shift to the PrPsc form, which would then aggregate. Exposure to the PrPsc form would then catalyze the conversion of normal PrPc to PrPsc. Hence, it would be transmissible by contact with just the PrPsc form of the protein. Likewise, species specificity could be explained if only PrPc dimers were present. PrPsc could form from proteins of the same species. The inherited form of the disease can be explained by the mutant form of the normal protein, which forms the beta structure more readily, leading to aggregation.
The same mutation in PrPc, Asp178Asn, can cause two distinct diseases: CJD and FFI. Which disease you get depends on whether you have 1 of two naturally occurring, nonharmful variants at amino acid 129 of the normal PrPc gene. If you have a Met at that position and acquire the Asp178Asn mutation, you get CJD. If, on the other hand, you have a Val at amino acid 129 and acquire the Asp178Asn mutation, you get FFI. This disease was first observed in 1986 and has been reported in five families worldwide. It manifests in the late 50s, and it affects men and women equally. It is characterized by a progressive loss of sleep and disrupted circadian rhythms. The brain shows neuronal losses. It is known that amino acids 129 and 178 occur at the start of alpha helices, as predicted from propensity calculations. Chronic exposure to micromolar levels of synthetic fragment 106-126 of PrPc kills hippocampal neurons. This peptide also has the greatest tendency to aggregate synthetic PrPc peptides.
Kuru killed many members of the Fore tribe in New Guinea until the cannibalistic practice of eating dead relatives was stopped. Analysis of the genes for the prion protein in the Fore tribe and other ethnic groups worldwide shows two versions differing by just one amino acid in all people (remember that a single gene is represented on both the maternal and paternal chromosomes). That these two forms exist worldwide suggests that they have been selected for by evolution and confer some biological advantage. People with just one form of the protein are more susceptible to developing prion diseases. Mead and Collinge have shown that about 75% of older Fore women (who had lived through cannibalistic practices) had two different prion genes, compared to about 15% of women from other ethnic groups. This high percentage suggests that these women were protected from the disease, leading, through natural selection, to a high proportion of heterozygotes in this defined population. The presence of two forms of the prion gene (which likely protect against prion disease) suggests that cannibalism might have been widespread among our early ancestors.
There appears to be a main difference between the formation of amyloid fibers from prion proteins and those from other proteins, such as mutant lysozymes. If you add mutant lysozyme to normal lysozyme, the amyloid fibers contain only the mutant protein. However, if you incubate mutant prion proteins with normal prions, the normal proteins become pathological.
Recently Added: 10/11/25
Polynucleotide Repeat Mutations
Deleterious protein aggregates can also occur in proteins whose gene contain anomalous expansions and repeats of triplet nucleotide-encoding key amino acids like glutamine and glycine. We will discuss two below.
Huntingtin Protein and Huntingon's Disease
A mutant version of the huntingtin protein causes the fatal neurological Huntington's Disease.
- The normal huntingtin gene has fewer than 35 CAG repeats (encoding a string of less than 35 polyQ in the protein). The polyQ section of the protein can form short intramolecular beta-strand structures that can interact with other huntingtin proteins to form some amyloid structure, but they may also stay extended.
- The huntingtin mutation causes more than 35 CAG repeats, leading to an expanded polyglutamine (polyQ) stretch in the N-terminal (encoded by the first exon of the HTTex1 gene) of the protein. This tract of polyGln forms a beta-hairpin connecting ~20-residue-long β-strands in a tight turn. These beta strands on one huntingtin can, through intramolecular hydrogen bonding, form amyloid fiber aggregates and undergo liquid–liquid phase separation.
The antiparallel β-sheets from the polyQ core in the mutant lack the usual twist found in amyloid fibers. Interactions between adjacent eight-residue glutamine strands are shown in Figure \(\PageIndex{xx}\) below.
 |
 |
Figure \(\PageIndex{xx}\). Structure of the polyQ beta structure hydrogen bonds in huntingtin. Bagherpoor Helabad, M., Matlahov, I., Kumar, R. et al. Integrative determination of the atomic structure of mutant huntingtin exon 1 fibrils implicated in Huntington's disease. Nat Commun 15, 10793 (2024). https://doi.org/10.1038/s41467-024-55062-8. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panel A: The eight-Gln building block used to generate polyQ core candidates. Six residues are shown faded out to bring the two non-faded in focus: these two are on adjacent β-strands of an antiparallel β-sheet, stabilized by backbone hydrogen bonds (purple), and continuous chains of side-chain hydrogen bonds (orange). The latter are crucial for packing the polar glutamines into the waterless amyloid core. When generating the core candidates, all χ1 and χ3 dihedral angles were independently rotated to explore all possible hydrogen-bond networks.
Panel E: Illustration of the inter-side-chain hydrogen-bond ladders (orange) in M2
The mutant polyQ fibrils form an amyloid core composed of many stacked β-sheets, with water excluded. Envision it as a series of glutamine "ladders" that form two distinct conformers, A and B. On either side of the polyQ in the primary sequence is an N17 (N-terminal end) and a proline-rich domain PRD at the C-terminal end. The PRD section is quite disordered and protects sites in the N17 region from post-translational modification (ubiquitination and phosphorylation), which could have led to protein degradation and reduced amyloid formation.
MATLEKLMKAFESLKSFQ44PPPPPPPPPPPQLPQPPPQAQPLLPQPQPPPPPPPPPPGPAVAEEPLHRP
Here are two models from molecular dynamics simulations of the aggregates. Figure \(\PageIndex{xx}\) shows an atomic model of the water-facing surface of the polyQ amyloid.

Figure \(\PageIndex{xx}\): Atomistic MD snapshot of the D2Q15K2 peptide fibril’s polyQ surface in contact with water. Exposed and buried Gln residues are colored green and gray, respectively. Note that the Gln side-chains within the amyloid core are well-ordered, while the water-facing side-chains exhibit greater mobility. Bagherpoor Helabad, M. et al., ibid
Figure \(\PageIndex{xx}\) shows a structural representation of the Q44-HTTex1 amyloid fibril.

Figure \(\PageIndex{xx}\): Structural representation of the Q44-HTTex1 amyloid fibril. Bagherpoor Helabad, M. et al., ibid
Panel A: A graphical depiction (after 5-μs simulation in Amber14SB86) of the HTTex1 fibril. The region shaded in gray denotes a single sheet within the fibril’s architecture.
Panel B: An atomic view of the N17 domain within the fibril, naming the specific amino acids.
Panel C: An atomic depiction of the glutamine side-chains within the fibril. The high stability of the fibril structure is primarily due to extensive hydrogen-bonding interactions among the glutamines, as depicted in the right panel.
Panel D: Top view representation of the β-sheet highlighted in panel (A). A quartet of HTTex1 monomers is visible. The polyQ is color-coded for the type “a” (red) and “b” (blue) strands; the tight β-turn is cyan. The N17 and PRD domains are orange and gray, respectively. Note the structural variation between different monomers in the same fibril sheet, including in particular the range of helical content in the N17 domain.
Download this amazing movie simulation of the Q44-HTTex1 fibril structure.
GGC repeat expansions and Fragile X-associated tremor/ataxia syndrome (FXTAS)
Expansions of another trinucleotide repeat, GGC, cause Fragile X-associated tremor/ataxia syndrome and neuronal intranuclear inclusion disease. The GGC repeats are translated into a polyGly sequence in protein when they are in the coding part of a gene, but can also be translated into different proteins if they are in the untranslated region near another site on the DNA. This non-canonical protein synthesis (translation) mechanism is called RAN (Repeat-Associated Non-AUG) translation. Transcription of the mRNA derived from the DNA sequence does not start with an AUG codon for the amino acid methionine.
Sometimes nucleotide repeats are 4 or 5 bases long. Given that translation is not regulated, multiple reading frames can be used to decode nucleotide repeats, allowing the synthesis of multiple proteins, including polyGly, polyAla, polyGln, polyPro, and polySer sequences.
PolyGly proteins (as well as the other poly sequences), given the small size of the side chain (-H), are inherently flexible and are very disordered. PolyGly and other polyAminoAcid proteins can form amyloid proteins, bind to important proteins (RNA-binding proteins, chaperones, etc), and generally disrupt RNA metabolism, leading to cell dysfunction and death. When genes for polyG proteins were engineered into cell lines, the resulting proteins formed long sequences that aggregated in the nucleus. They also recruited other proteins naturally enriched in glycine. The human protein with the most glycines is FAM98B, a subunit of the enzyme tRNA ligase complex (tRNA-LC). This is used to splice together tRNA exons after intron removal. When polyG proteins were expressed, they bound FAM98B, leading to nonfunctional tRNA-LC in the nucleus. In mice, this cause severe neurological disorders in the brain.
To be added after resolution of the Government Shutdown: FAM98B: AF-Q52LJ0-F1: undefined. https://www.uniprot.org/uniprotkb/Q5...ntry#sequences and iCn3D model
Misfolding and Aggregation Summary
Recent work has shown that proteins considered harmless can form misfolded intermediates that aggregate into pre-fibril structures, which are toxic to cells. This process is usually prevented in the cell by the interaction of nascent protein forms with chaperones, which sequester exposed hydrophobic patches and prevent aggregation. (Obviously, prion proteins and the others mentioned above are exceptions.) Amyloid fibers (characterized by subunits with an abnormal amount of beta-structure) can be made from many different types of proteins, as noted above. Is this property specific to just a handful of proteins, or is it more common than expected from the limited examples noted so far? The new studies show that when a bacterial protein, HypF, is incubated at pH 5.5 in the presence of trifluoroethanol, aggregates (but not fibrils) form with enhanced beta structure. These aggregates slowly form into fibrils characteristic of amyloid protein fibers. The early aggregates (formed before fibril formation) were cytotoxic. Similar results were seen with dimers and trimers (prefibril states) of the amyloid-β peptide released from cultured neurons.
A diverse group of proteins that do not share significant secondary or tertiary structures can form amyloid-like protein aggregates. Even though their monomer forms share little in common, the insoluble amyloid aggregates have a common structure in which the monomers within the aggregates exhibit significant beta structure, with the strands running perpendicular to the aggregate axis. Since it has recently been shown that almost any protein, under the "right" set of conditions, can form such aggregates, the stabilizing feature of protein aggregates may be present in any protein. Evidence suggests that the polypeptide backbone, and not the side chains, is key in forming stable interstrand H-bonds in beta-secondary structures in amyloid aggregates. In contrast, native, nonamyloid forms of normal proteins arise through specific interactions between unique side chain sequences and structures, which outcompete nonspecific interactions among backbone atoms found in amyloid structures. Nonspecific aggregation becomes more prevalent when buried hydrophobic side chains and main chain atoms become more solvent-exposed. Such exposure occurs when native proteins form intermediate molten globule states under altered solvent conditions or when destabilizing mutants of the wild-type protein arise. Some mutations may alter protein folding cooperativity, thereby increasing the fraction of non-native protein states. Other mutations that decrease the protein's charge or increase its hydrophobicity might enhance aggregation. In addition, chemical modifications to proteins (such as oxidation or deamination) may destabilize the native state, leading to the formation of the molten globule state. Once formed, this state may aggregate through sequestration of exposed side-chain hydrophobes or inter-main-chain H-bond formation. Aggregate formation appears to proceed through the initial formation of soluble units (which may or may not be more toxic to cells than the final aggregate). Aggregates are kinetically stable species. Since amyloid aggregates are cytotoxic and almost any protein can form them, albeit with different propensities, nature, through evolutionary selection, has presumably disfavored proteins with high tendencies to form such aggregates.
Accurate protein folding is required for cell viability. Aberrant protein folding can clearly cause serious illness. Given the extraordinary nature of the task and its failure, the process governing protein folding must be highly regulated. Figure \(\PageIndex{20}\) shows the steps determining intracellular concentrations and locations of normal and aberrant protein structures.

Potential therapies for proteostasis diseases include replacing aberrant proteins, shifting the equilibrium toward active forms with small ligands, or modulating pathways with agents that influence processes such as signal transduction, transcription, translation, degradation, and translocation. This can be achieved using molecules such as siRNAs to modulate the levels of chaperones, disaggregases, and signaling pathways.
Recently updated: 10/3/25
Functions of Amyloid Proteins in Microbes
Are all amyloid proteins toxic? It depends on the cell and your reference frame. It turns out that both bacteria and fungi can form biofilms. These are beneficial to them, but not to infected humans, as these films are often resistant to drug treatment. Biofilms are aggregates, or perhaps better, communities of microorganisms that bind to a surface and encapsulate the enclosed cells within an extracellular matrix (ECM). The films can stick to almost any surface, including rocks, plant and animal tissue, pipes, teeth, and medical implants. If you ever walked on a dock near a lake and felt you were on ice, you were likely "skating" on an algal biofilm. Amyloid proteins from the cells in the biofilm are critical for biofilm formation and structure. They are involved in the initial attachment of cells and in cell:cell interactions. The cells in the biofilm are protected from the effects of environmental chemicals, including antibiotics and immune regulators. They are also resistant to dehydration and pH changes. As biofilms protect internal cells, the reversible conversion between dispersed ↔ enclosed cells is highly regulated. For example, in E. coli, the transcription factor CsgD leads to the synthesis of cellulose and a fibrous structure called the Curli fimbriae, which contain bacterial amyloid proteins. The amyloid protein forms aggregates enriched in β-sheets, which then form fibrils.
An example of a fungal amyloid protein is the Heterokaryon incompatibility protein S (HetS) from the filamentous fungus Podospora anserina. Fungi can reproduce sexually by releasing spores, which can grow filamentous hyphae as part of their life cycle, as illustrated in Figure \(\PageIndex{21}\). The figure shows two spores fusing to form a hyphal network, which, in this case, leads to the growth of a recognizable mushroom.

Figure \(\PageIndex{21}\): Fungi reproduction. Fungi_sessuate_reproduction.png: M.violante. https://commons.wikimedia.org/wiki/F...production.png.
There are two forms of the HETS protein from two different alleles: Het-s, which can exist in both a soluble "normal" form and a prion amyloid version, and Het-S, a soluble form of the protein. Het-S has a few amino acid differences in its N-terminus that prevent it from adopting a prion form on its own.
What prevents two different strains from fusing? HetS is involved. If one strain expresses Het-s and the other Het-S, the strain with Het-s induces cell death in the Het-S strain. The het-s prion interacts with and induces a prion form of Het-S, which leads to conformational changes in Het-S to generate an amyloid β-solenoid form. In addition, the N-terminal of Het-S changes conformation to expose a hydrophobic region that interacts with membranes. At the membrane, Het-S forms amyloid oligomers that puncture it, leading to cell death.
Figure \(\PageIndex{22}\) shows interactive iCn3D models of the predicted AlphaFold structure of the soluble form of Het-s (Q03689) and the C-terminal amino acids 218-289 prion form (2LBU). The prion aggregate contains five monomers containing amino acids 218-289 of the protein, and is bound to Congo Red, a dye that binds to amyloid fibers.
|
Unavailable during US Government shutdown. AlphaFold model of the HET-s soluble monomer (AF-Q03689-F1). (Copyright; author via source). Click the image for a popup or use this external link: |
NMR structure of the HET-s amyloid (multimer bound to Congo Red. (2LBU). (Copyright; author via source). Click the image for a popup or use this external link: https://www.ncbi.nlm.nih.gov/Structu...9cbf00612c5921 |
.jpg?revision=1&size=bestfit&width=343&height=200)
.jpg?revision=1&size=bestfit&width=343&height=222)
As mentioned earlier, biofilms are beneficial to microorganisms, but not humans. Recently, the mouth bacteria viridans streptococci, which are found in dental biofilms, were found in about 60% of coronary artery plaques in autopsies of people who died suddenly. Since the bacteria were in biofilms, they would be resistant to antibiotics and immune mediators. In heart attacks, the plaques rupture, likely releasing the bacteria, which are then recognized by the immune system. This leads to a burst of immune activity from the innate (macrophages) and adaptive (T-cell) immune systems. The sequestration of bacteria in coronary artery biofilms might explain the limited effect of antibiotics in treating cardiovascular disease. These findings link oral health and heart attacks, and suggest that antibiotics might be useful at the onset of a heart attack.
One last twist in the case of biofilms. Helicobacter pylori is a stomach bacterium found in a high percentage of people. It has been associated with ulcers and gastric cancer. The CagA protein (Cytotoxicity-associated immunodominant antigen) produced by the bacterium is a virulence factor. It's injected into epithelial cells that line the gut. Once in the cell, it alters morphology and signaling, leading to the loss of contact inhibition in epithelial cells. This can promote tumor formation, making the protein a bacterial oncoprotein. The N-terminal of the protein, CagAN, is involved in amyloid protein binding.
However, the secreted form has several advantages from both the bacterial and human perspectives. It binds to both amyloid fibril proteins of bacteria (curli, CsgA, E. coli) and FapC (Pseudomonas) with high affinity and also human amyloid proteins (Aβ42/Aβ40 peptides and tau in Alzheimer’s disease, and α-synuclein in Parkinson’s disease), inhibiting their fibril formation. It can prevent biofilm formation in bacterial cells and destabilize existing ones. Binding again is high affinity. It affects different stages of amyloid fiber formation for different cell types, as illustrated in Figure \(\PageIndex{23}\) below.

Figure \(\PageIndex{23}\): CagAN interferes with different microscopic events of the fibril formation process of various amyloid peptides or proteins.
Zhen Jin et al. Helicobacter pylori CagA protein is a potent and broad-spectrum amyloid inhibitor. Sci. Adv.11,eads7525(2025).DOI:10.1126/sciadv.ads7525. https://creativecommons.org/licenses/by-nc/4.0/
The kinetics of amyloid aggregation involve distinct stages: In the primary nucleation phase, monomers come together to create a nucleus (kn), from which a fibril can start to elongate (k+); simultaneously, during secondary nucleation (k2), monomers adhere to the fibril’s surface, catalyzing the development of a new nucleus and facilitating exponential fibril growth. Refer to the schematic representation in (A) and (B) for a visual depiction of this process. (A) CagAN suppresses amyloid formation of different bacterial functional amyloid proteins, i.e., CsgA from E. coli and FapC from Pseudomonas. For these amyloidogenic proteins, CagAN predominantly blocks elongation, as indicated by a red cross. (B) CagAN impedes the formation of amyloid fibrils by various human pathogenic amyloid peptides or proteins through diverse mechanisms. In the case of AD-associated Aβ42 and Aβ40 peptides, primary nucleation is largely blocked, as CagA proteins likely stabilize oligomers. Conversely, for the AD-relevant protein tau, both secondary nucleation and elongation are affected by CagAN. Regarding the PD-related protein α-synuclein, CagA inhibits elongation. The IAPP peptide, associated with T2D, is inhibited from fibrillizing by CagAN, affecting both secondary nucleation and elongation.
Figure \(\PageIndex{24}\) below shows an AlphaFold 3 model of the interaction of GagAN and amyloid proteins. GagAn has three domains, domain I (D1), domain II (D2), and domain III (D3).

Figure \(\PageIndex{24}\): AlphaFold 3 models of the interaction of GagAN and amyloid proteins. Zhen Jin et al., ibid.
AlphaFold 3 prediction of the interactions of CagAN and different amyloid peptides or proteins, with four Aβ42 subunits mimicking the aggregates (left) and six α-synuclein mimicking the fibrils.
Binding, Intracellular Granules and Droplets
The above structures are fascinating aggregates of specific proteins. The aggregates are quite large. In Alzheimer's Disease, they vary from around 150-500 μm2, giving a length of 12-22 μm if they were squares. In comparison, intracellular "granules" are much smaller, with diameters of 200-500 nm (0.2-0.5 μm). The term "granule" describes particles in cells that are barely visible by light microscopy. Granules are found in many cells and mostly contain protein. Platelet granules contain many proteins involved in clotting. Pancreatic beta cell granules contain insulin for secretion. Other types of granules in germ-line cells are dense bodies, perinuclear P granules in Caenorhabditis elegans, germinal granules in Xenopus laevis, chromatoid bodies in mice, and polar granules in Drosophila. It contains both RNA and proteins. These are often referred to as ribonucleoprotein (RNP) granules. Plants and livers also contain starch granules (a carbohydrate). The granules don't appear to be surrounded by a membrane. Rather, they are just aggregates of proteins or of RNA and proteins. Chapters 10 and 11 show analogous particles for lipids, nonpolar "insoluble" molecules that self-aggregate into micelles and membrane bilayers. In contrast to the granules mentioned above, lipid droplets containing TAGs and cholesterol esters are surrounded by a phospholipid monolayer with adsorbed protein. Understanding the structure and properties of phase-separated granules may provide insight into the aggregates formed in neurodegenerative diseases.
How do these granules form? What principle underlies the specificity of proteins and RNAs found in them? The aggregates are not toxic, unlike the beta-amyloid aggregates discussed above. Granule formation can be caused by a classic "phase transition/separation", not unlike gaseous water, which can self-associate through hydrogen bonds to form liquid drops, which can freeze with the formation of more hydrogen bonds to form solids. Soluble biomolecules in cells can reversibly aggregate through multiple, weak noncovalent interactions to form storage granules. This balance might be perturbed if storage granules aggregate further, potentially through a process that is irreversible and has health consequences, as observed in neurodegenerative diseases. Let's delve into new insights into the processes involved in droplet formation.
Imagine small amounts of a sparingly soluble oil added to an aqueous solution. Initially, it is in solution, but at higher concentration, dipole-dipole interactions, along with the “hydrophobic effect,” would drive the oil out of solution into liquid drops. This phase separation, also known as liquid-liquid demixing, occurs when two liquids (solubilized oil in water and separated oil drops) separate. This process has produced various types of non-membrane-bound droplets (distinct from membrane-bound vesicles) within the cell.
Figure \(\PageIndex{25}\) below shows the many types of condensates found in cells.

Figure \(\PageIndex{25}\): Condensates found in cells. PHASE SEPARATION 101. Animation Lab. Margot Riggi, Janet Iwasa, et al. Creative Commons Licensing CC BY 4.0 - https://creativecommons.org/licenses/by/4.0/
This phenomenon has also been seen with intrinsically disordered proteins and proteins with such domains. These are characterized by amorphous structures composed of repeated, often positively charged amino acids and contain a limited number of distinct amino acids. An example of a protein with a domain that has low sequence complexity is the SP1 transcription factor, a DNA-binding protein. One of its transactivation domains has almost 20% glutamines, with regions within it having even higher percent abundances. It has been estimated that up to 20% of eukaryotic proteins lack a stable shape, as they are, in part, intrinsically disordered and contain low-complexity domains (LCDs). They are found in the N- and C-terminal ends of all mammalian intermediate filament proteins, as well as in almost all RNA-binding proteins, lining the nuclear pore, and on the cytoplasmic faces of mitochondrial, lysosomal, peroxisomal, and Golgi integral membrane proteins. They are the target of up to 3/4s of post-translational modifications. LCDs, therefore, appear to facilitate promiscuous binding of various proteins, especially those that add or remove covalent tags.
Under the right conditions, these can aggregate and “precipitate” from the solution. What is the nature of the precipitate? It may have properties more akin to distinct liquid droplets, so that this process could be referred to as liquid-liquid demixing.
Properties of demixed drops include reduced rates of material diffusion into and out of the drop, coupled material movements within the drop, and probable weak hydrophobic-dependent aggregation, which makes drops sensitive to agents such as detergents. Liquid-like diffusion inside the drop is observed, as evidenced by the rapid fluorescence recovery from partially photobleached internal components in the drop.
As with forming a crystalline solid from a liquid solution, the process must be initiated by seeding. This process can be “catalyzed” by poly (ADP-ribose), a nucleic acid-like polyanion that interacts with intrinsically disordered proteins. The negative charges would counterbalance the positive charges in the disordered protein domain, which, without neutralization, would interfere with protein-protein contacts necessary for aggregation/droplet formation and demixing. Aggregation in these cases may arise from hydrophobic interactions (even though hydrophobic side chains are underrepresented in the disordered domains).
- A database, PhaSePro, is dedicated to proteins that drive liquid-liquid phase separation (LLPS) in living cells.
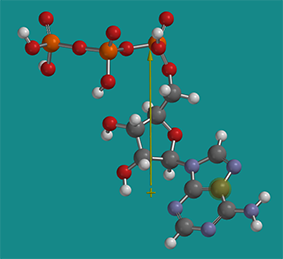
The solubility of proteins in cells is a fascinating topic in its own right. A high ATP concentration (5 mM) in the cell facilitates protein solubilization. ATP is considered a hydrotrope. It’s a small molecule with a distinct polar part (polyphosphate and ribose) and a more nonpolar part (the adenosine ring). Hence, it acts like a mini-detergent (an amphiphile), but it doesn’t form micelles. It helps stabilize more nonpolar regions of proteins in solution and has been shown to inhibit aggregate formation and disaggregate preexisting aggregates. Figure \(\PageIndex{26}\) shows a protonated form of energy-minimized ATP with its dipole moment as an arrow from the + to - ends. The dipole moment would only be larger if the ATP was deprotonated and had negative charges.
Biochemists also use the term gel (examples include polyacrylamide gel or fibrin blood clots, which are chemically cross-linked) and a "gel" form of a bilayer (Chapter 10), when they wish to describe a structure that is neither solid nor liquid. Structures like the cytoskeleton or the actin-myosin network would be examples of the latter.
Noncovalent gels would be characterized by the regulatable dissociation of subunits and, hence, short half-lives. A gel (either covalent or noncovalent) with a high water content is called a hydrogel, as it contains hydrophilic components. An example is RNA- and protein-containing particles.
Here is a link to Phase Separation 101, a wonderful description of phase separations in cells by Janet Iwasa and the Animation Lab. It contains numerous animations illustrating phase separations in chemical and cellular systems, and it describes the underlying chemical and physical processes.
Granules
Granules that contain RNA and proteins are called ribonucleoprotein bodies (RNPs) or RNA granules. Specific examples include cytoplasmic processing bodies, neuronal and germ granules, and nuclear Cajal bodies, nucleoli, and nuclear dots/bodies. Some granules contain proteins, including inclusion bodies with misfolded and aggregated proteins, as well as those with active proteins involved in biosynthesis, such as purinosome (for purine biosynthesis) and cellulosomes (for cellulose degradation).
The presence of trinucleotide repeats in DNA and RNA can lead to the aggregation of RNAs and proteins encoded by genes containing the repeats. We saw above examples of proteins with extended stretches of glycine and glutamine that aggregate. What about expansions of a charged amino acid like arginine? It likely would not aggregate with itself given charge-charge repulsion. More likely, it would interact with a polyanion, like RNA or DNA.
Arginine (Arg, R) is encoded by six codons: CGU, CGC, CGA, CGG, AGA, and AGG. As with the cases of Gly and Gln, runs of Arg occur using the codons CGG, AGA, and AGG. Short stretches of Arg (5-20) can normally occur, and are used as signals to translocate a translated protein into the nucleus and also ot bind to RNA. These functional proteins do not aggregate abnormally. However, proteins produced by RAN synthesis from expanded repeats can form aberrant proteins that damage cells. One example is the RAN synthesis from the hexanucleotide repeats of GGGGCC (G4C2) found in the first intron the protein C9orf72 gene, resulting in a protein hghly enriched in arginine.
This gene is the most common genetic cause of Lou Gehring’s Disease or amyotrophic lateral sclerosis (ALS) and also of frontotemporal dementia (FTD). This gene lacks an AUG start codon and can be read to produce five different dipeptide repeat (DPR) proteins, the most common being poly-GR and poly-PR, both of which are highly toxic. Since they have a high positive charge, they are likely disordered. They bind readily to RNA (negatively charged) and nucleoli. Hence, they impact transcription, splicing, and the formation of the membraneless organelle, the nucleolus, which is the site of ribosome formation for normal protein synthesis. They can lead to phase separation of RNA and other RNA-binding proteins, forming stress granules. They are toxic even at low levels. In Fragile X syndrome, 230-4000 repeats of the CGG triplet occur in the noncoding parts of the genome, compared to less than 50 in the normal gene.
Trinucleotide expansion occurs in the non-protein-coding intronic DNA, which also leads to trinucleotide repeats in the RNA spliced out of the transcribed RNA. For example, a CTG DNA repeat in an intron leads to a poly (CUG) RNA, which, through unusual base pairing, can form RNA aggregates, as seen in myotonic dystrophy.
In vitro experiments show that small complexes are soluble. Still, as the size increases, liquid-liquid demixing (or a liquid-gel transition) can occur, forming spherical droplets of RNA particles. This would explain the observation that pathologies occur above a certain repeat length. If misfolded proteins are also present, these particles might combine to form larger gels.
In the control experiment, demixing and spherical particle formation were not observed when the repeats were scrambled. In an experiment similar to adding 1,6-hexanediol to intrinsically disordered proteins, small antisense trinucleotide repeats, such as (CTG)8, which could interfere with the weak H bonds between G and C in the aggregates, were added. The size of RNA drops (foci) was reduced. In vivo experiments showed characteristic drop-like structures only when the repeats were sufficiently large.
Researchers found that in vitro, RNA drop formation was inhibited by monovalent cations. In the presence of 0.1 M ammonium acetate, which permeates cells without affecting pH, CAG RNA droplets in vitro disappeared.
Aggregation of mRNA may be one way to regulate its translation and, in turn, indirectly regulate gene activity. There are advantages to regulating the translation of a protein from mRNA, especially if the mRNA's "activity" can be dynamically regulated. This would be useful if new protein synthesis were immediately required. Hence, reversible aggregation is one way to regulate mRNA activity (in addition to degradation).
Protein drops and granules
The cytoskeletal proteins actin and tubulin (a heterodimer of alpha and beta chains) can exist in either soluble or condensed, filamentous states (as actin filaments and microtubules, respectively). GTP hydrolysis is required for the formation of tubulin polymers. Actin binds ATP, which is necessary for filament formation, but ATP cleavage is required for depolymerization. Hence, nucleotide binding/hydrolysis regulates the filament equilibrium, unlike simple phase changes in water.
Since only certain proteins form granules, they must have similar structural features that facilitate reversible binding interactions. These proteins have multiple, weak-binding sites, but if they act collectively, they provide multivalent (multiple) binding interactions that allow robust but not irreversible granule formation. Here are some characteristics of proteins found in granules:
- the protein NCK has three repeated domains (SH3) that bind to proline-rich motifs (PRMs) in the protein NWASP. These proteins are involved in actin polymerization. In high concentrations, they precipitate from the solution and coalesce to form larger droplets;
- repeating interaction domains are widely found, especially among RNA-binding proteins;
- some proteins contain Phe-Gly (FG) repeats separated by hydrophilic amino acids in portions of the protein that are intrinsically disordered.
- a biotinylated derivative of 5-aryl-isoxazole-3-carboxyamide (Figure \(\PageIndex{27}\)) precipitates proteins, which are enriched in those that bind RNA (RBPs). The precipitated proteins were generally intrinsically disordered and characterized by low-complexity sequences (LCS). One such example contained 27 repeats of the tripeptide sequence (G/S)Y(G/S). The proteins could also form hydrogels (made of hydrophilic polymers and crosslinks) and transition between the soluble and gel phases, with extensive hydrogen-bond networks. The hydrogel gel phase exhibited an X-ray diffraction pattern similar to that of beta-structure-enriched amyloid proteins. Short-range, weak interactions between LCS may then drive reversible condensation into gel-like granule states, characterized by extensive hydrogen bonding (similar to that during ice formation). If this process goes awry, more continued and irreversible formation of a solid fibril (as seen in neurodegenerative diseases) might occur from the hydrogel state.

- RNAs form granules when proteins bind to them via RNA-binding domains that interact via low-complexity sequences, leading to phase separation and the formation of hydrogel-like granules. Around 500 RNA-binding proteins have been found in the human RNA interactome. They are enriched in LCSs and have more tyrosines than average proteins in the entire proteome, in which tyrosines are often found in the (G/S)Y(G/S) motif. Phosphorylation of tyrosines (Y) in LCS may decrease association and hydrogel stability.
Given that many neurodegenerative diseases are associated with unfolded/misfolded protein aggregates, the high protein concentrations in protein-containing liquid drops may pose cellular problems. If sufficiently high, the equilibrium might shift from a liquid drop to a solid precipitate, with severe cellular consequences. The progression to the solid state may irreversibly affect the cell.
Low complexity domains (LCD) and neurodegenerative disease
The aggregation of alternatively folded proteins is associated with neurodegenerative disease. Mutations that cause disease are associated with the formation of low-complexity domains and aggregates, a phenomenon increasingly described as phase separation. The demixed phases are stabilized by interchain backbone hydrogen bonds, as shown in the many beta-sheet aggregates described above. Evidence suggests that labile structures with the potential for interchain H-bonds and beta-strand formation lead to fibril formation. The nascent interactions would involve short stretches of interchain H bonds. If so, mutations that enrich such nascent structural interaction would promote fibril formation, while those that inhibit the nascent interactions would inhibit fibril formation. A study (Zhou et al, Science, 377, 2022. DOI: 10.1126/science.abn5582) verifies this.
The investigators created single amino acid variants of the low-complexity domains of an RNA-binding protein, TDP-43 RNA, that prevented the single amino acid within a region involved in interchain beta strand formation from forming a hydrogen bond via its amide hydrogen. They achieved this by methylating the single main-chain amide nitrogen, which prevents its participation in hydrogen bonding. The modification is shown in Figure \(\PageIndex{28}\).

Figure \(\PageIndex{28}\): Methylation of a single backbone nitrogen in a region involved in interchain hydrogen bond and beta sheet formation in the low complexity domain of proteins
Of the 23 variants they made, 9 were within a continuous stretch, inhibiting phase separation. As determined by cryo-EM, these nine were at the same sites as hydrogen bonds between adjacent chains of the TDP-43.
Next, they looked at other proteins with low-complexity domains that form aggregates/polymers. The proteins examined were the neurofilament light (NFL) chain protein, the microtubule-associated tau protein, and the heterogeneous nuclear RNA-binding protein A2 (hnRNPA2). They identified 10 mutations in LCDs that are known to be associated with neurological diseases. Indeed, these mutations allow for an extra hydrogen bond in low-complexity domain sequences and enhance aggregate formation, presumably through the extra interchain H bond. Specifically, the known mutations replace individual prolines. This cyclic amino acid lacks an amide hydrogen and, therefore, cannot donate a hydrogen bond to another amino acid, which allows for one additional hydrogen bond. Each known mutation was associated with neurological disease and increased stable aggregate/polymer formation. This increased aggregation/polymer (phase separation) was reversed in vitro by chemical methylation of the single amino acid change in the mutant, which prevented it from forming hydrogen bonds. Site-specific methylation was performed by linking synthetic peptides containing the single Nα-methyl amino acid to other synthesized peptides that comprise the protein. The semisynthetic NFL protein, for example, was incubated under conditions conducive to the assembly of mature intermediate filaments.
In vitro experiments were conducted using different synthetic head domains of the neurofilament light (NFL) chain protein, in which the P8 residue contained a different amino acid at that position. Figure \(\PageIndex{29}\) shows the variant amino acids.

Figure \(\PageIndex{29}\): Variant amino acids used at position P8 in the low complexity head domain of the neurofilament light (NFL) chain protein (after Zhou et al, ibid)
Only variants containing Leu at position P8 could form filaments, as measured in vitro using fluorescent assays. Experiments like this are crucial in determining whether phase separation/aggregation causes are causative or merely correlated with the development of complex neurodegenerative diseases.
Summary
Protein aggregation is a critical phenomenon that occurs when certain proteins, rather than folding into a single, unique native conformation, adopt alternative, often misfolded structures that self-assemble into larger, insoluble aggregates. Although many proteins naturally form functional oligomers and filaments through specific, reversible interactions, a subset of proteins—including prions and amyloid-forming proteins—can undergo a drastic conformational change that transforms predominantly α-helical or random coil structures into β-sheet–rich forms. This conversion exposes new hydrogen-bonding surfaces that drive the templated, self-propagating formation of fibrils.
Such amyloid fibrils are characterized by a common cross-β architecture, in which β-strands from individual monomers align perpendicularly to the fibril axis and are stabilized by extensive intermolecular hydrogen bonding. Although the monomeric forms of these proteins may appear benign, the aggregated state is associated with several devastating neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and prion diseases. In these conditions, the aggregates can interfere with normal cellular functions, ultimately leading to cell death.
Research has revealed that the aggregation process is not random; it can be influenced by specific amino acid sequences, point mutations, and post-translational modifications that either promote or inhibit the conversion to the aggregated state. Moreover, while intracellular protein quality control mechanisms—such as molecular chaperones and the unfolded protein response—normally prevent harmful aggregation by assisting correct folding and targeting misfolded proteins for degradation, these systems can become overwhelmed under stress conditions or due to genetic mutations.
Recent structural studies using techniques such as cryo-electron microscopy and NMR have provided detailed models of amyloid fibrils from various proteins, revealing surprising similarities in their cross-β structures despite differences in the native states of the precursor proteins. In addition, emerging evidence on phase separation reveals that some cellular condensates and RNA granules form through reversible, weak multivalent interactions—a process that, when dysregulated, may also contribute to pathological aggregate formation.
Overall, this chapter emphasizes the delicate balance between protein folding, aggregation, and cellular proteostasis. It highlights how understanding the molecular mechanisms of aggregation—from initial misfolding events to the formation of toxic fibrils—can inform therapeutic strategies to prevent or reverse protein aggregation in neurodegenerative diseases.