
7: Enzymes and Kinetics
- Page ID
- 158572
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Define Gibbs Free Energy (ΔG) and explain its role in determining reaction spontaneity.
- Differentiate between exergonic and endergonic reactions.
- Explain how ATP hydrolysis can drive non-spontaneous reactions through energy coupling.
- Understand how environmental factors (e.g., temperature, concentration) influence ΔG.
- Relate spontaneity (thermodynamics) to enzyme catalysis (kinetics).
- Describe the basic structure of enzyme-catalyzed reactions.
- Define key rate constants involved in enzyme kinetics (k₁, k₋₁, k₂).
- Understand the concept of the enzyme-substrate complex and its role in catalysis.
- Interpret the meaning of the dissociation constant (Kd) and its relationship to binding affinity.
- Recognize the relevance of enzyme binding affinity in drug design (e.g., Imatinib for BCR-ABL).
- Apply the Michaelis-Menten equation to describe and predict enzyme behavior.
- Analyze enzyme efficiency using Km and Vmax.
- Interpret kinetic data using a Lineweaver-Burk plot.
- Differentiate between competitive and non-competitive enzyme inhibition.
- Connect kinetic parameters (Km, Vmax, Kd, k₁) to real-world clinical and pharmacological scenarios.
- Explain how enzyme kinetics guide the design and evaluation of drugs (e.g., HIV protease inhibitors, acetaminophen metabolism, doxycycline absorption).
- What are the steps in a typical enzyme-catalyzed reaction?
- How does enzyme binding strength affect its function and potential as a drug target?
- Why is the reversibility of substrate binding important?
- ΔG (Gibbs Free Energy): A thermodynamic value that predicts if a reaction is spontaneous.
- Exergonic Reaction: A reaction that releases energy (ΔG < 0); spontaneous.
- Endergonic Reaction: A reaction that requires energy input (ΔG > 0); non-spontaneous.
- ATP Hydrolysis: The breakdown of ATP to ADP + Pi, releasing energy (ΔG ≈ –30.5 kJ/mol).
- Coupling Reactions: Linking an unfavorable (endergonic) reaction with a favorable one to make it proceed.
- Activation Energy (ΔG‡): The energy barrier that must be overcome for a reaction to proceed.
- Enzyme Catalyst: A biological molecule that speeds up reactions by lowering activation energy.
- Le Chatelier’s Principle: When a system is disturbed, it shifts to minimize the disturbance.
- Vmax: The maximum velocity of an enzymatic reaction when the enzyme is saturated with substrate.
- Km: Substrate concentration at half-maximal velocity (Vmax); reflects binding affinity.
- Enzyme (E): A biological catalyst that speeds up chemical reactions.
- Substrate (S): The molecule upon which an enzyme acts.
- Enzyme-Substrate Complex (ES): A transient complex formed when an enzyme binds its substrate.
- k₁: The rate constant for the forward reaction forming ES.
- k₋₁: The rate constant for the reverse reaction, dissociating ES into E and S.
- k₂ (or kcat): The rate constant for conversion of ES to product (P).
- Kd (Dissociation Constant): A measure of enzyme-substrate affinity; calculated as k₋₁ / k₁. A lower Kd indicates stronger binding.
- Affinity: The strength of binding between an enzyme and its substrate.
- Catalysis: The acceleration of a chemical reaction by a catalyst.
Spontaneity of a Reaction and Gibbs Free Energy (ΔG)
In chemistry, the term spontaneity refers to whether a reaction can proceed on its own, without needing continuous input of external energy. This is not about speed but about thermodynamic favorability. The primary tool for determining spontaneity is the Gibbs Free Energy change, denoted as ΔG. If ΔG is negative, the reaction releases energy into the surroundings and is called exergonic—this means the reaction is spontaneous and will proceed naturally. On the other hand, if ΔG is positive, the reaction requires energy input, making it endergonic, and thus non-spontaneous under standard conditions.
- If ΔG is negative, the reaction releases energy—this is an exergonic reaction and is spontaneous.
- If ΔG is positive, the reaction requires energy input—this is an endergonic reaction and is non-spontaneous.
To illustrate this, consider a biological example: the conversion of glucose into glucose-6-phosphate, an essential first step in glycolysis. This reaction has a ΔG° of +18 kJ/mol, indicating that it is non-spontaneous (Figure 7.1). This is similar to trying to roll a ball uphill; the system needs an input of energy to push it forward. Cells, however, need this reaction to happen regularly as part of their energy metabolism. To drive this uphill reaction forward, nature uses a clever trick called energy coupling. The cell pairs this unfavorable reaction with a highly favorable one: the hydrolysis of ATP (adenosine triphosphate) into ADP (adenosine diphosphate) and inorganic phosphate (Pi). ATP hydrolysis has a ΔG° of –30.5 kJ/mol, meaning it releases a significant amount of energy—like a ball rolling down a steep hill. By combining the two reactions—glucose phosphorylation (+18 kJ/mol) and ATP hydrolysis (–30.5 kJ/mol)—the cell effectively merges their energy changes. The net ΔG° becomes –12.5 kJ/mol, making the overall reaction spontaneous. In simpler terms, this is like trying to buy a $50 item when you only have $20. You can’t afford it—unless you also have a $30 gift card. Combining your money and the gift card, you now have $50, enough to buy the item. That’s what happens in the cell: ATP acts like an energy currency or a gift card that allows you to “pay for” processes that wouldn’t occur otherwise.
This concept of G-coupling is fundamental in biochemistry because many essential reactions—like protein synthesis, active transport of ions, and DNA replication—are inherently endergonic and would not proceed on their own. Cells rely on the free energy released from ATP hydrolysis or other high-energy compounds to push these processes forward. In this way, coupling allows biological systems to achieve thermodynamically unfavorable outcomes by linking them to favorable reactions. Without this mechanism, the machinery of life would come to a halt.
Example:
Consider this reaction (Figure 7.2):
- Glucose → Glucose-6-Phosphate, with ΔG° = +18 kJ/mol
- This is a non-spontaneous reaction under standard conditions. It won’t proceed without external energy.
- ATP → ADP + Pi has ΔG° = -30.5 kJ/mol.
- Nature often drives non-spontaneous reactions forward by coupling them with highly spontaneous reactions, such as ATP hydrolysis.
- Glucose + ATP → Glucose-6-Phosphate + ADP
- The ΔG° of the coupled reaction = +18 (from glucose phosphorylation) + (–30.5 from ATP hydrolysis) = –12.5 kJ/mol
Now, the net ΔG° is negative, so the overall reaction becomes spontaneous. This is how biological systems make “unfavorable” reactions proceed—by borrowing energy from ATP or other high-energy compounds.

Figure 7.1: Reaction from Glucose to Glucose-6-Phosphate.

Figure 7.2: ΔG coupling reaction of Glucose to Glucose-6-Phosphate
Factors That Affect Reaction Spontaneity
The spontaneity of a chemical reaction—whether it can proceed on its own—is determined by the Gibbs Free Energy change (ΔG). But ΔG itself can be influenced by several environmental and molecular factors. One important factor is the nature of the molecules involved. Some chemical bonds are inherently unstable and prone to breaking, while others are very stable. For instance, the breakdown of hydrogen peroxide (H₂O₂) into water and oxygen occurs spontaneously because the O–O bond in peroxide is weak and easily broken. In contrast, breaking the C–H bonds in methane requires a lot of energy, making the reaction non-spontaneous unless conditions are adjusted.
Environmental conditions such as temperature, pressure, and pH also affect ΔG. According to the Gibbs free energy equation (ΔG = ΔH – TΔS), increasing the temperature (T) can make a reaction more favorable if it increases entropy (ΔS). That’s why some reactions that are non-spontaneous at room temperature can become spontaneous when heated. For example, melting ice is non-spontaneous below 0°C, but it is spontaneous above it due to entropy increasing as solid turns into liquid.
Concentration of reactants and products is another key factor. According to Le Chatelier’s Principle, increasing the concentration of reactants shifts the equilibrium to favor product formation, effectively making a reaction more favorable under certain conditions. For instance, in glycolysis, the accumulation of glucose and inorganic phosphate (Pi) helps push the formation of glucose-6-phosphate, even if the standard ΔG° is positive.
Lastly, and perhaps most powerfully, is the strategy of reaction coupling. As we’ve discussed, energetically unfavorable reactions (positive ΔG) can be “paid for” by coupling them to very favorable reactions (negative ΔG), such as ATP hydrolysis. This is a fundamental strategy in cells. Without it, many essential biosynthetic and metabolic processes could not proceed
-
Nature of the Molecules – Some bonds naturally break or form more easily due to inherent chemical stability.
-
Conditions – Temperature, pressure, and pH can shift ΔG. High temperatures often make endergonic reactions more feasible.
-
Concentration – High concentration of reactants can push reactions forward via Le Chatelier’s Principle.
-
Coupling Reactions – As explained, unfavorable reactions can piggyback on favorable ones to proceed.
While ΔG tells us if a reaction can occur, it tells us nothing about how fast it will occur. That’s where reaction kinetics and the rate constant (k) come into play. The rate constant is a numerical value that reflects how quickly a reaction occurs under a given set of conditions. It’s influenced by factors like temperature, molecular orientation, and activation energy.
According to collision theory, for a reaction to happen, reactant molecules must collide. But not just any collision will do—the molecules must strike each other with the correct orientation and sufficient energy to overcome the activation energy barrier (ΔG‡) (Figure 7.3). This energy barrier represents the "hill" the reactants must climb before they can be converted into products. The rate constant (k) increases when this barrier is lower or when collisions happen more frequently and effectively.

Figure 7.3: Collision Reaction for a substrate to become a product.
There are three main ways to increase the rate of a reaction:
-
Increase substrate concentration: Imagine a crowded room where more people are likely to bump into each other. Similarly, more substrate molecules mean a higher probability of effective collisions, which increases reaction rate.
-
Increase temperature: Heating the system makes molecules move faster, increasing the number and energy of collisions. This boosts the chances that collisions will overcome the activation barrier.
-
Add enzymes: Enzymes are biological catalysts that don't just increase the rate of reactions—they do so with incredible precision. Enzymes help orient the reactant molecules properly and stabilize the transition state, effectively lowering the activation energy (ΔG‡). Think of an enzyme as a skilled dance partner who guides your movements perfectly so that the dance (reaction) happens smoothly and efficiently.
A dramatic illustration of how enzymes affect reaction rates is seen with the enzyme urease, which catalyzes the breakdown of urea into ammonium carbonate. Without urease, this reaction occurs extremely slowly—just one reaction every 3 million years. That’s practically geological time! But with urease, the reaction rate skyrockets to about 30,000 reactions per second. This is because urease drastically lowers the activation energy (ΔG‡) required, making the reaction occur almost instantly under biological conditions. This example demonstrates the enormous power of enzymes—not to change ΔG, or spontaneity, but to change the speed at which equilibrium is reached. They allow life to perform thousands of chemical reactions in a controlled and timely fashion. Without enzymes, metabolism would grind to a halt.
- The activation energy (ΔG‡) is the energy needed to get reactants into a transition state, an unstable intermediate on the way to forming products (Figure 7.4).
- Even if a reaction has a negative ΔG (spontaneous), it might proceed slowly if ΔG‡ is high.
- Enzymes lower ΔG‡, not ΔG, to speed up reactions.
- ΔG: Energy difference between reactants and products (predicts spontaneity).
- ΔG‡: Energy needed to reach the transition state (predicts rate).

Figure 7.4: ΔG shows the energy difference between the reactant and the product, while ΔG‡ is the Gibb free energy of transitional state.
Enzyme Specificity and Binding Models
One of the most fascinating features of enzymes is their specificity—the ability to select and act on a particular substrate among many similar molecules. This is crucial in the body because it ensures that reactions occur in a controlled and precise manner. For example, the enzyme lactase specifically acts on lactose, the sugar found in milk, and breaks it down into glucose and galactose. If lactase acted on many sugars indiscriminately, our metabolism would be chaotic and inefficient.
Enzymes can also show stereospecificity, meaning they only interact with one enantiomer (mirror image) of a molecule. A common example is how enzymes in our bodies typically recognize and process L-amino acids, not D-amino acids, which are rarely found in nature. This is highly relevant in drug design: some drugs have both L and D forms (called chiral drugs), but only one form is effective or safe. The painkiller ibuprofen, for example, exists in two enantiomers, but only the S-enantiomer is active—this shows the importance of enzyme stereospecificity in how drugs work.
There are two primary models used to explain how enzymes bind to substrates: the Lock and Key model and the Induced Fit model (Figure 7.5). In the Lock and Key model, the enzyme has a rigid active site that matches the substrate’s shape exactly, like a key fitting into a lock. This model explains high specificity, as only the correct “key” (substrate) will fit. A classic example is the enzyme acetylcholinesterase, which breaks down the neurotransmitter acetylcholine—the active site of this enzyme is so specific that it won’t act on structurally similar molecules. However, most enzymes follow the Induced Fit model, which is more dynamic. In this model, the enzyme changes shape slightly to better accommodate the substrate once it starts to bind. This explains how enzymes can stabilize the transition state—the unstable, high-energy structure that forms during a reaction. A real-life example is hexokinase, an enzyme that adds a phosphate group to glucose during glycolysis. When glucose binds, hexokinase undergoes a conformational change that “clamps down” on the substrate, creating a snug fit and lowering the activation energy needed for the reaction.
Enzymes are highly specific and may recognize:
- Specific substrates
- Specific stereoisomers (e.g., only L-amino acids)
Binding models:
- Lock and Key: Substrate fits enzyme perfectly (like a key in a lock).
- Induced Fit: Enzyme molds around the substrate to fit better—this is more common and explains how enzymes stabilize the transition state.

Figure 7.5: (A) Lock and key enzyme model and (B) induced fit enzyme model. Image is shared under a CK-12 license and was authored, remixed, and/or curated by CK-12 Foundation via source content that was edited to the style and standards of the LibreTexts platform.
Cofactors and Coenzymes
While many enzymes can function with just their amino acid side chains, others require additional non-protein components to carry out their catalytic activity. These helpers are broadly classified into cofactors and coenzyme (Figure 7.6).
Cofactors are typically inorganic ions, such as magnesium (Mg²⁺), zinc (Zn²⁺), iron (Fe²⁺), or heme groups. These ions often help in stabilizing charges during the reaction or are directly involved in electron transfer. For instance, the enzyme carbonic anhydrase, which helps convert carbon dioxide and water into carbonic acid in your blood, uses zinc as a cofactor to hold water molecules in the correct position and polarize them for the reaction. Another example is cytochrome P450, a heme-containing enzyme family in the liver that helps detoxify drugs and toxins—many common drugs, including statins and antidepressants, are metabolized by this system.
Coenzymes, on the other hand, are organic molecules, often derived from vitamins, that act as temporary carriers of atoms or electrons during enzymatic reactions. For example, NAD⁺ (nicotinamide adenine dinucleotide) and FAD (flavin adenine dinucleotide) are derived from vitamin B3 (niacin) and vitamin B2 (riboflavin), respectively. They serve as electron carriers in redox reactions, accepting and donating electrons as needed. A classic example is alcohol dehydrogenase, an enzyme in the liver that helps break down ethanol (alcohol). This enzyme uses NAD⁺ to accept electrons from ethanol, converting it into acetaldehyde, a toxic intermediate that is further processed into acetic acid and eventually eliminated. This process is crucial in detoxifying alcohol from the body. Another well-known coenzyme is Coenzyme A (CoA), derived from vitamin B5 (pantothenic acid). CoA is essential for transferring acyl groups, especially in fatty acid metabolism. For example, during the breakdown of fats for energy, fatty acids are converted to fatty acyl-CoA before entering the mitochondria. This step is essential for producing ATP from fats.
Understanding enzyme specificity, binding models, and the role of cofactors/coenzymes is not just textbook knowledge—it’s directly applicable to real-world biology and medicine. For instance, many enzyme inhibitors are designed as drugs to block specific enzymes. ACE inhibitors (like lisinopril) block the angiotensin-converting enzyme, reducing blood pressure. Their effectiveness relies on the drug mimicking the natural substrate of the enzyme, fitting into the active site based on these binding principles. Similarly, vitamin deficiencies can impair enzyme function because they limit coenzyme availability. A deficiency in vitamin B3, for example, results in pellagra, which manifests with symptoms like diarrhea, dermatitis, and dementia because many redox reactions in energy metabolism require NAD⁺.
Enzymes often need helpers to function:
- Cofactors: Inorganic (e.g., Zn²⁺, Fe²⁺, Mg²⁺, or heme groups)
- Coenzymes: Organic molecules, often vitamins or derivatives (e.g., NAD⁺, FAD, CoA)
- Example: Alcohol dehydrogenase uses NAD⁺ to remove electrons from ethanol during detoxification.
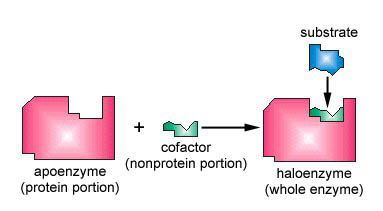
Figure 7.6: An apoenzyme and cofactor combine to form a haloenzyme. If the cofactor is an organic molecule, it is called a coenzyme. Image is shared under a CC BY 4.0 license and was authored, remixed, and/or curated by Gary Kaiser via source content that was edited to the style and standards of the LibreTexts platform.
Enzyme Kinetics and Function
Enzyme kinetics is the study of how fast enzymatic reactions occur and what factors influence their speed. At the core of this understanding is a simplified reaction model involving an enzyme (E), a substrate (S), an enzyme-substrate complex (ES), and the product (P). The reaction begins with the enzyme binding the substrate to form the ES complex. This step is governed by the forward rate constant k1. However, this binding is reversible, meaning the complex can also fall apart back into the enzyme and substrate, and this backward step is governed by the rate constant k−1. Once the ES complex is formed, the reaction can proceed to convert the substrate into product, releasing the enzyme to act again. This final step is governed by another rate constant, k2, which reflects the speed at which the product is formed from the ES complex.
Basic enzyme kinetics reaction:

- k₁ = rate constant for formation of ES from E + S
- k₋₁ = rate constant for dissociation of ES back into E + S
- k₂ = rate constant for the conversion of ES into product (P)
- We often assume no product is present initially, so k2 can be ignored in early analysis.
At the very beginning of a reaction, when almost no product has accumulated yet, we often simplify the kinetics by ignoring the reverse conversion of product back to ES. This allows us to focus on how effectively the enzyme binds the substrate and converts it into a product. One important concept that arises from this is the dissociation constant, denoted as Kd which is calculated as Kd=k-1/k1=[E][S]/[ES]. This equation describes the equilibrium between free enzyme and substrate versus the bound complex. A low Kd value means the ES complex is very stable, indicating that the substrate binds tightly to the enzyme (Figure 7.7). Conversely, a high Kd value implies weak binding and a tendency for the complex to fall apart easily. This concept of Kd is critical when designing or evaluating drugs. For example, if a drug is designed to inhibit an enzyme involved in cancer cell growth, such as a kinase like BCR-ABL in chronic myeloid leukemia, the drug must bind very tightly to be effective. Imatinib (Gleevec) is a famous example of a drug that works because it has a very low Kd for BCR-ABL, allowing it to stay bound for a long time and block the enzyme’s function. In contrast, a drug with a high Kd would quickly fall off the enzyme and require higher doses or might not be effective at all.
Dissociation Constant (Kd)

- Low Kd = Tight binding (substrate sticks well, strong affinity)
- High Kd = Weak binding (substrate doesn’t bind well)
- Example: A drug with low Kd stays bound longer and may be more effective.
Building on this, enzyme kinetics also looks at how the velocity of the reaction changes depending on how much substrate is present. This is captured by the Michaelis-Menten equation: V0=[Vmax][S]/[Km+S]. This equation expresses the initial velocity of the reaction (V0) as a function of substrate concentration ([S]). Vmax is the maximum rate at which the enzyme can work, and Km is the substrate concentration at which the reaction velocity is half of Vmax. The value of Km gives us a sense of how much substrate is needed to efficiently engage the enzyme (Figure 7.8). If Km is low, it means the enzyme reaches half-maximal speed with a small amount of substrate, indicating high affinity. If Km is high, more substrate is needed to get the enzyme working effectively, which points to low affinity. Consider enzymes involved in glucose metabolism. Hexokinase, which operates in most tissues, has a very low Km for glucose, meaning it binds glucose tightly and works well even when glucose levels are low. This is important in the brain and muscles, where glucose is vital. Glucokinase, found in the liver, has a much higher Km, meaning it only becomes active when glucose is abundant—after a meal, for example. This distinction allows the liver to store excess glucose without stealing it from more glucose-sensitive organs.
Michaelis-Menten Kinetics

- Vmax = Maximum velocity (when all enzymes are saturated)
- Km = Substrate concentration at which reaction is at half Vmax
- Low Km = Strong affinity (needs less substrate to reach half-max speed)
- High Km = Weak affinity

Figure 7.7: kd is the dissociation constant at equilibrium.

Figure 7.8: Km defines the affinity of substrates.
The relationship between substrate concentration and reaction velocity creates a curved graph—a hyperbola—when you plot V0 against [S]. But this shape can make it difficult to accurately calculate Vmax and Km (Figure 7.9). That’s why biochemists use a transformed version of the equation, called the Lineweaver-Burk plot, which is simply a double reciprocal of the Michaelis-Menten equation (Figure 7.10). In this format, the graph plots 1/V0 on the Y-axis versus 1/[S]on the X-axis, resulting in a straight line. The slope of this line is Km/Vmax, the Y-intercept is 1/Vmax, and the X-intercept is −1/Km. This linear transformation makes it easier to determine the kinetic constants from experimental data.
Lineweaver-Burk plot

This is the same as y = mx + b (a straight line):
- y-axis = 1/V₀
- x-axis = 1/[S]
- y-intercept = 1/Vmax
- x-intercept = –1/Km
In practical terms, these kinetics are used to evaluate how drugs interact with enzymes. If a new HIV protease inhibitor is being tested, researchers can use the Michaelis-Menten equation to determine how the drug affects Km and Vmax. A competitive inhibitor will raise the apparent Km (because it competes with the substrate for binding), but won’t change Vmax. A non-competitive inhibitor, on the other hand, doesn’t affect binding affinity (Km remains the same), but it reduces Vmax, because it prevents the enzyme from working even when the substrate is bound. Understanding enzyme kinetics in this way—through reaction rates, binding constants, and saturation behaviors—is fundamental not only to enzymology but also to pharmacology, biotechnology, and clinical biochemistry. Whether you're designing a drug, understanding a metabolic pathway, or evaluating enzyme deficiencies, these principles provide a detailed and powerful framework to interpret biological reactions.

Figure 7.9: Michaelis-Menten graph.

Figure 7.10: Lineweaver-Burk plot and its equation
Understanding the Application of Kinetics
Let's walk through each different examples and explain the enzyme kinetics in detail, so you can clearly understand the concepts like Vmax, Km, and rate constants (k₁) with real-world context.
Example 1 - Understanding Vmax
- Hexokinase is an enzyme responsible for phosphorylating glucose to form glucose-6-phosphate—the very first step in glycolysis, which is the primary pathway cells use to extract energy from glucose. Different isozymes of hexokinase function in different tissues, and here, let's compare two: Hexokinase A with a high Vmax, and Hexokinase B with a low Vmax. Vmax refers to the maximum rate of reaction when the enzyme is saturated with substrate. A high Vmax means the enzyme can process a large number of glucose molecules quickly when glucose is abundant. Therefore, Hexokinase A is more active, especially under high glucose conditions, because it can work at a faster rate. Conditions that affect Vmax include the total concentration of enzyme present and whether the enzyme is saturated. Vmax does not depend on how tightly the enzyme binds to the substrate (that’s Km) but rather how quickly it can catalyze the reaction once the substrate is bound. So if your body expresses more of Hexokinase A, or if you're in a tissue like muscle that demands fast glucose metabolism, you'll have a more "active" glucose metabolism.
- Imagine that glucose molecules are customers and hexokinase enzymes are cashiers. Hexokinase A (high Vmax) is like a super-fast cashier—they can scan and check out a lot of customers quickly. Hexokinase B (low Vmax) is a slower cashier—they still process everyone, but not as quickly. So, if there’s a big rush of customers (high glucose), the fast cashier (Hexokinase A) is more efficient and clears the line quickly. This means Hexokinase A is more active, especially when there’s lots of glucose. Conditions affecting Vmax would be like the number of cashiers you have. If you hire more (increase enzyme concentration), the total checkout speed (Vmax) increases
Example 2 - Understanding Km
- Alcohol dehydrogenase (ADH) is the enzyme that breaks down alcohol into acetaldehyde, a toxic compound that is then further broken down into harmless acetate. The Km of an enzyme tells you how much substrate (in this case, alcohol) is needed for the enzyme to work at half its maximum speed. A low Km means the enzyme has high affinity—it binds alcohol tightly and can start breaking it down even at low alcohol concentrations. Here, the normal enzyme has a Km of 0.746 mM, while Mutant A has a higher Km (1.5 mM) and Mutant B has a lower Km (0.2 mM). Since Mutant B binds alcohol most tightly, it is more active at low alcohol concentrations. Mutant A, with the highest Km, is the least efficient and would require higher alcohol levels to function effectively. This directly ties into the Asian Glow phenomenon. Many East Asians have a variant of alcohol dehydrogenase (or the downstream enzyme ALDH) that either has altered Km or Vmax, leading to inefficient metabolism of acetaldehyde. This causes it to build up, leading to facial flushing, nausea, and rapid heart rate after drinking even small amounts of alcohol.
- Think of alcohol as a Velcro ball and the enzyme as a Velcro paddle. A low Km is like a paddle with very sticky Velcro—the ball (alcohol) sticks easily, even when there's not much of it. A high Km is like a paddle with worn-out Velcro—you need to throw more balls (higher alcohol concentration) before any of them stick. Thus, Mutant B (Km = 0.2 mM) is the stickiest (best binding), whereas Mutant A (Km = 1.5 mM) barely catches any balls—it’s the weakest.
Example 3 - Understanding K1
- Acetaminophen (Tylenol) is a common pain reliever that is generally safe at normal doses. It is metabolized by the liver, and a small portion is converted into a toxic metabolite called NAPQI. Normally, your body neutralizes this toxin quickly through a process involving glutathione. However, alcohol consumption increases the k₁ rate constant of liver enzymes, meaning the enzyme binds to acetaminophen faster and shunts more of it into the toxic pathway. This leads to higher levels of NAPQI. Alcohol also depletes the liver’s glutathione reserves, further reducing the body's ability to detoxify NAPQI. As a result, if you take acetaminophen after drinking alcohol, you risk severe liver damage because the enzymes are working faster (higher k₁), more toxin is formed, and less is neutralized. That’s why it’s strongly recommended not to take acetaminophen after drinking or during a hangover.
- Imagine your liver is a factory with conveyor belts that handle acetaminophen. Normally, it processes the drug slowly and safely. Alcohol turns up the speed (increases k₁), making the conveyor belt run much faster. Now, more drug is being processed quickly, but some of it takes a toxic side route. With the belt moving faster, more drug is accidentally shunted to this dangerous path. Meanwhile, alcohol has also used up your safety nets (glutathione). The result? Toxic buildup and potential liver damage. That’s why drinking alcohol before taking acetaminophen is dangerous—it speeds up the wrong reaction pathway.
Sub Example 4 - Low K1
- Doxycycline is an antibiotic that is also used in lower doses to treat acne due to its anti-inflammatory properties. It has a high k₁, meaning it is absorbed quickly and efficiently when taken on an empty stomach. However, this absorption is sensitive to interactions with metal ions, such as calcium, magnesium, and iron found in milk and multivitamins. When you consume milk or vitamins close to your doxycycline dose, the metal ions bind to the drug and form a complex—a process called chelation. This complex cannot be absorbed through the intestinal wall, effectively lowering the k₁ and making the drug much less bioavailable. In practice, this means doxycycline may not work as intended, and bacteria or acne might not be treated effectively. That’s why it’s recommended to avoid milk and supplements for at least a couple of hours before and after taking doxycycline.
- Think of doxycycline as a letter trying to be mailed (absorbed into the body). The digestive system is like a mailbox that only accepts plain envelopes. If you drink milk or take vitamins, the metal ions act like duct tape, wrapping around the envelope. Now the mailbox (your body) can’t accept it, and the letter just sits there. The chelation lowers the effective k₁ (rate at which doxycycline enters the bloodstream). So, the drug doesn’t get absorbed properly and won’t work well.
Example 5 - High K1
- Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID) commonly used for pain and inflammation. It has a very high k₁, meaning it is absorbed rapidly into the bloodstream when taken orally. However, this rapid absorption can come with a cost—it can irritate the stomach lining, especially when taken without food. When you take ibuprofen on an empty stomach, it reaches the stomach wall quickly and at high concentrations. Since ibuprofen also inhibits COX-1, an enzyme that protects the stomach lining by producing prostaglandins, this leads to reduced mucus protection and increased acid damage. As a result, the risk of ulcers, bleeding, and irritation increases significantly. Taking ibuprofen with food doesn’t reduce the drug’s effectiveness much but slows down its absorption (lowers effective k₁) and provides a buffer that protects the stomach lining. That’s why doctors often recommend eating something before taking ibuprofen, especially for people who use it regularly.
- Think of it this way: Ibuprofen is like hot sauce—effective but intense. Your stomach lining is like bare skin. If you eat food first, it acts like a shirt that protects your skin. The hot sauce still gets absorbed (just a bit slower), but the stomach doesn’t feel the burn. But if you take ibuprofen on an empty stomach, there’s no protection. The hot sauce goes straight on the skin, causing irritation, ulcers, or even bleeding. That’s why food buffers and slows absorption (reduces the punch of k₁) to protect you.
- In your own words, how does ΔG differ from activation energy (ΔG‡)? Which tells you about speed, and which tells you about favorability?
- Why is ATP often called the “energy currency” of the cell?
- What would happen in a cell if endergonic reactions could not be coupled with ATP hydrolysis?
- How do enzymes affect the activation energy of a reaction, and how does this relate to urease’s role in catalyzing urea breakdown?
- Why does increasing temperature make some non-spontaneous reactions become spontaneous?
- Imagine you are developing a drug to block an enzyme that catalyzes an unfavorable reaction in cancer cells. Would you target the ΔG of the reaction or the enzyme’s active site? Why?
- A student argues: “Since the phosphorylation of glucose has a positive ΔG, it can't happen in cells.” How would you respond using the concept of coupling?
- How does Le Chatelier’s Principle help push forward a reaction with a small positive ΔG?
- Think about how drugs like ibuprofen (a COX enzyme inhibitor) or statins (HMG-CoA reductase inhibitors) work by exploiting enzyme kinetics and specificity.
- Revisit glycolysis. Why is it important that some steps are coupled to ATP hydrolysis?
- How does an enzyme’s Km value reflect its efficiency in different tissues or organisms?
- Why is it dangerous to mix alcohol and acetaminophen from a kinetic perspective?
- If a new drug is found to increase the k₁ of an enzyme reaction, what might be the implications?
- How could changes in enzyme kinetics (e.g., in alcohol dehydrogenase) explain variable patient responses to alcohol?