
4.3: M13 Phage Display Libraries
- Page ID
- 18142

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Microbial systems developed for experimental molecular evolution
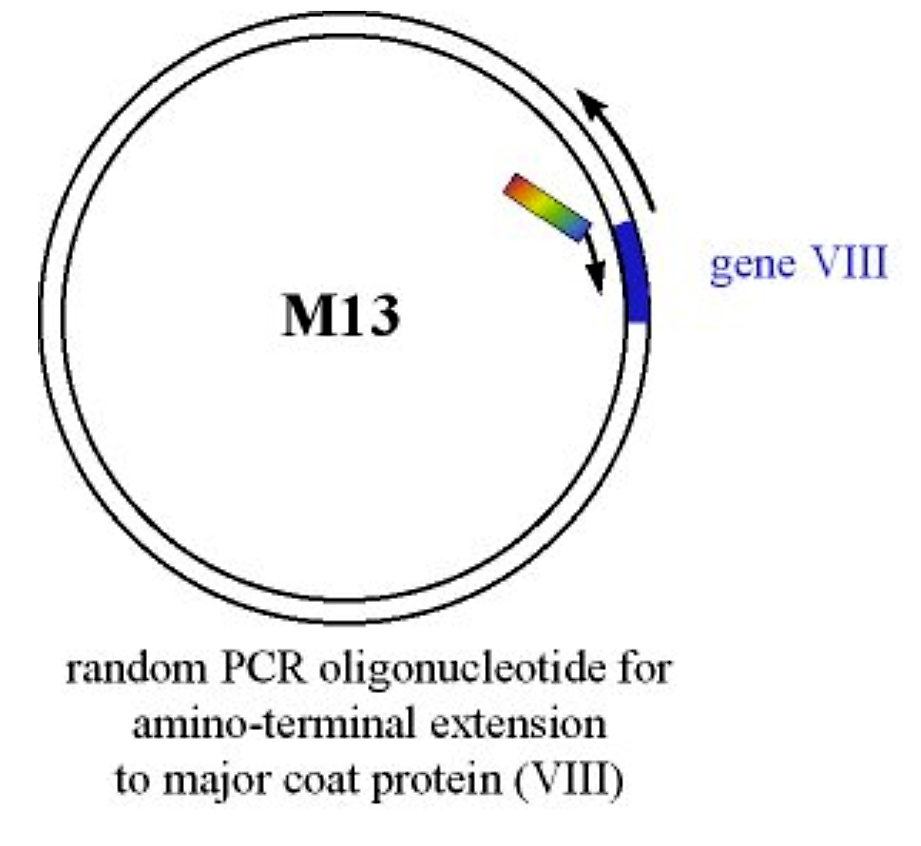
A protein or peptide is introduced at the amino-terminus of the major coat protein (Gene VIII) via PCR-based mutagenesis
.png?revision=1&size=bestfit&width=405&height=369)
Figure 4.3.1: Introduction of protein at gene VIII
The linear PCR product is amplified to regenerate an intact circular viral genome whose gene VIII protein now has an amino terminal extension. The mutagenized M13 is introduced into E. coli cells. It is naked DNA (not packaged with viral coat proteins) so it is introduced into E. coli like a plasmid (i.e. via electroporation or using CaCl2 competent E. coli cells).
- The extruded viral progeny will:
- Be somewhat longer as the genome is larger but should be packaged o.k.
- Express the mutant gene VIII (major coat) protein as part of the filamentous phage assembly
.png?revision=1&size=bestfit&width=363&height=417)
Figure 4.3.2: Mutated M13 progeny
Selection
The amino-terminal peptide may have affinity for a specific ligand, possibly an antibody. We can use this binding affinity to purify the mutant phage. We can can construct a chromatography column where the chromatography matrix has attached to it the ligand of interest. If we pass our phage through this column, the mutant phage containing the mutant gene VIII protein will bind to, and be immobilized, on the column
.png?revision=1&size=bestfit&width=448&height=325)
Figure 4.3.3: Immobilized ligand
- Any wild-type phage will not bind, and will pass through the column
- The bound phage can be eluted and used directly to reinfect E coli to propagate viruses with the mutant gene VIII protein
The mutant gene VIII protein which has been purified carries along with it the DNA which codes for itself. In other words, we have specifically selected for the DNA which codes for the gene VIII of interest (i.e. the mutant)
Changing binding specificity
Suppose we have a slightly different ligand attached to the matrix…
.png?revision=1&size=bestfit&width=168&height=95)
Our phage with mutant gene VIII protein will not bind very well to this different ligand. If we introduce the appropriate amino acid changes into our mutant gene VIII protein, we may be able to improve the binding affinity (specificity) and get the phage to bind to the new ligand
.png?revision=1&size=bestfit&width=461&height=423)
- After PCR and ligation, this will produce a library of phage genomes, each with a potentially different sequence for the amino terminal extension of the gene VIII protein
- Generally speaking, when these are introduced into E. coli the efficiency is typically so low that, at most, each successful transformation results in only one phage genome per E. coli
- Progeny phage from our library will, therefore, have only one type of gene VIII protein per phage (a single phage will have the gene VIII protein which it's DNA codes for).
- If we pass the collection of progeny phage from our library over our ligand column, we will selectively bind those phage whose mutant gene VIII protein has affinity for the ligand
.png?revision=1&size=bestfit&width=586&height=420)
Figure 4.3.5: Specific binding
- If we isolate the bound phage we can re-infect E.coli, and isolate the progeny phage
- In this first selection we may have many other phage which might be weak, or generally non-selective, ligand binders. So, the process is repeated until we have a library of specific high-affinity mutants (this is sometimes called "Bio-mining" or "Bio-panning")
- We can then sequence the DNA and find out what sequence(s) in the gene VIII mutant provide high binding specificity to our new ligand
Practical limitations
- Over what length of DNA/protein can we randomize?
Example: a random stretch of 10 amino acids
- 2010 possible different unique polypeptide sequences. Each amino acid is 110 g/mol or 1.8 x 10-22g per amino acid. This would mean that a complete random library of 10mer peptides would have a mass of 1.8 x 10-9 g.
Example: a random stretch of 20 amino acids
- 2020 possible different unique polypeptide sequences. Each amino acid is 110 g/mol or 1.8 x 10-22g per amino acid. This would mean that a complete random library of 20mer peptides would have a mass of 18.9 Kg!
This does not even consider the significantly larger mass of the virus particle of which it would be a part. This type of calculation demonstrates that generating random DNA sequence libraries is limited to very short stretches of about half a dozen or so codon positions.
Random Mutagenesis
We could randomly introduce all four bases at each position (NNN) while trying to synthesize a mutagenic random codon, but what would this give us?
| Leu, Arg, Ser (6 codons/64) | 9.4% |
| Val, Pro, Thr, Ala, Gly (4 codons/64) | 6.3% |
| Ile (3 codons/64) | 4.7% |
| Phe, Tyr, His, Gln, Asn, Glu, Asp, Cys (2 codons/64) | 3.1% |
| Met, Trp (1 codon/64) | 1.6% |
| Stop (3 codons/64) | 4.7% |
- Thus, not all amino acids are present in equal amounts, and we will have 4.7% stop codons
- We can improve this by using all four bases at the first two positions, and an equal mix of G and C bases at the third (wobble) position. This would result in 32 possible codons and the following distribution of amino acids and codons:
| Phe, Tyr, Cys, Trp, His, Gln, Ile, Asn, Met, Lys, Asp, Glu, Stop (1 codon/32) | 3.1% |
| Gly, Val, Ala, Pro, Thr (2 codons/32) | 6.3% |
| Leu, Arg, Ser (3 codons/32) | 9.4% |
Another general problem with the above method is that of non-specific binding
- This is due in part to the large number of mutant gene VIII proteins (~2500) on the virus surface.
- Ligand binding may be due to the interaction of two or more gene VIII proteins contributing to binding the ligand (i.e. "polyvalent" binding)
- In this case, the isolated mutant peptide may show weak or no binding affinity at all
Approach #1 - Mutate gene III protein instead of gene VIII
- While there are ~2,500 copies of the gene VIII (major coat protein) per phage, there are 5 copies of the gene III (minor coat protein) per phage
.png?revision=1&size=bestfit&width=291&height=358)
Figure 4.3.6: Mutating gene III
- We can introduce the mutagenic sequence to the amino-terminal of the gene III protein and thereby limit the number of copies on the phage particle
- this may still result in polyvalent binding
Approach #2 - Supplement with wild-type gene III protein during phage assembly
- If the M13 phage contains a second copy of gene III which we do not mutate then when the phage assembles (as it extrudes through the outer membrane) the five gene III proteins it picks up will be a mixture of mutant and wild type (in roughly equal amounts). In this case, the progeny phage will have, on average, 2-3 copies of the mutant gene III protein
- If we can increase the expression of the wild type gene III protein, to the point where it is present in four-fold excess over the mutant, then the progeny phage will, on average, contain 1 copy of the mutant gene III protein (and ligand binding will, by definition, be mono-valent)
- We can put a strong promoter in front of the wild type gene III
- We could place the wild type gene III near one of the terminators (after gene IV, for example). This position would result in higher expression of the wild type gene due to the cumulative effects of the upstream promoters
Variation
Expression of mutagenic protein as a fusion of an outer membrane protein of E.coli itself
- We can attach our mutagenic sequence to a gene for an outer membrane protein (e.g. OmpT, LamB)
- The surface of the E. coli cell itself displays the mutagenic peptide
- Outer membrane proteins may be present at a level of 10,000 molecules or more
- Our ligand column will selectively bind specifically binding E. coli cells. These can be eluted and cultured and the process repeated to select for the mutant sequence with binding affinity
Expression of mutagenic protein as a fusion with a DNA binding protein
- We can attach our mutagenic sequence to a gene for a protein which binds DNA (e.g. lac repressor)
- The mutagenic lac repressor will be encoded by a plasmid which itself contains two lac operator sequences
- The mutant repressor molecule expressed by the plasmid will bind strongly to the lac operator regions on the very same plasmid
- The cells are lysed (the repressor remains bound to its associated plasmid DNA) and the ligand binding step is performed to enrich for ligand binding sequences
- The associated plasmid DNA is isolated and transformed back into E. coli for amplification and another round of selection
.png?revision=1&size=bestfit&width=607&height=421)
Figure 4.3.7: Fusing mutagenic protein and binding protein