
28.9: Gated Ion Channels - Neural Signaling
- Page ID
- 14995


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)-
Understand the Fundamental Role of Ion Channels in Neural Signaling:
- Explain how ion channels facilitate the movement of ions across the hydrophobic membrane.
- Differentiate between constitutively open (leakage) channels and gated channels that open or close in response to stimuli such as ligands, changes in voltage, or mechanical forces.
-
Describe the Structure and Functional Organization of Neurons:
- Identify the key structural components of neurons (cell body, dendrites, axon, synapses) and their roles in signal transmission.
- Compare and contrast neuromuscular junctions with central nervous system synapses, emphasizing differences in neurotransmitter release and receptor activation.
-
Analyze the Biophysical Basis of Membrane Potential Generation and Maintenance:
- Describe how ion gradients, created and maintained by ion pumps (e.g., Na⁺/K⁺-ATPase) and selective permeability through channels, contribute to the resting membrane potential.
- Interpret how differential ion permeabilities (as demonstrated by synthetic vesicle studies versus biological membranes) influence the electrochemical potential across cell membranes.
-
Explain the Mechanisms Underlying the Action Potential:
- Outline the sequential changes in ion permeability (Na⁺, K⁺, Cl⁻) that produce the phases of depolarization, repolarization, and hyperpolarization in an action potential.
- Evaluate the impact of voltage-gated ion channels in generating rapid electrical signals essential for neuronal communication.
-
Differentiate Between Ionotropic and Metabotropic Neurotransmitter Receptors:
- Define ionotropic receptors and describe how they directly gate ion flow upon neurotransmitter binding, resulting in rapid synaptic transmission.
- Define metabotropic receptors and discuss how they indirectly modulate ion channel activity via G-protein-coupled second messenger cascades, leading to slower, modulatory effects.
-
Assess the Impact of Neurotoxins on Ion Channel Function:
- Identify key neurotoxins (e.g., tetrodotoxin and saxitoxin) and explain their modes of action in blocking specific ion channels.
- Discuss how these toxins help reveal the structural features and selectivity mechanisms of ion channels.
-
Evaluate How Neurotransmitter Binding Influences Neuronal Excitability:
- Explain how binding of excitatory neurotransmitters (e.g., acetylcholine, glutamate) and inhibitory neurotransmitters (e.g., GABA) alters membrane potential.
- Describe how changes in membrane potential can trigger or prevent the generation of an action potential.
-
Integrate the Roles of Ion Channels and Neurotransmitter Receptors in Synaptic Transmission:
- Discuss how the combined activities of ligand-gated and voltage-gated channels determine the overall synaptic response.
- Explain the importance of synaptic integration in neural networks, including how single neurons can process multiple excitatory and inhibitory signals.
-
Relate Neural Signaling to Higher-Level Brain Functions:
- Appreciate the complexity of the human brain by relating the molecular mechanisms of neural signaling to processes such as memory, cognition, and behavior.
- Consider how alterations in ion channel function and neurotransmitter signaling can contribute to neurological disorders.
-
Apply Quantitative and Structural Approaches to Neural Signaling:
- Interpret experimental data and computational models (e.g., electrophysiological recordings, ion permeability measurements) to explain how ion fluxes generate membrane potentials.
- Use structural models of ion channels and receptors to understand their function and specificity in neural signaling.
These learning goals will help students build a comprehensive understanding of the molecular and biophysical underpinnings of neural communication and its significance in health and disease.
Introduction to Neural Signaling
Ion channels are membrane proteins that allow the flow of normally impermeant ions across the hydrophobic "sea" of cell or organelle membranes. The channel proteins can be constitutively open or "gated" open (or closed) by signals that affect the conformation of the protein. The signals can be ligands (in the form of hormones or neurotransmitters), post-translational modifications (mostly phosphorylation, dephosphorylation), or more difficult-to-understand events (change in transmembrane potential, pressure, or temperature). Gated channels are perhaps best studied through their role in neural signaling.
Neurochemistry is one of the most explosive areas of biological research. Scientists are unraveling the molecular bases for memory, cognition, emotion, and behavior. The next decades will bring a greater understanding of brain chemistry, along with it the potential to alter human mood and memory, and to treat mental illnesses such as schizophrenia much more effectively. The human brain has about 86 billion neurons. Compare this with estimates for the number of stars in the Milky Way galaxy (about 200 billion, derived from luminosity and mass measurements, but some estimates suggest 400 billion). Now imagine that each neuron can form connections - synapses - with 1000 to 10,000 other neurons. Throw in another 86 billion nonneuronal cells, like glial cells, and you have one of the most complex structures in the universe. A recent high-resolution map of the cerebral cortex shows that a single cubic millimeter of the brain contains 57,000 cells, about 150 million synapses (connections between cells), and about 0.2 meters of blood vessels. This section will explore the biology and chemistry of neurons.
Recent studies suggest around 3300 different types of cells in the brain. We will discuss two kinds of neurons - those that interact with muscles at the neuromuscular junction and those that interact with other neurons in the central nervous system. Neurons consist of a single, nucleated cell body with multiple signal-receiving outgrowths (dendrites, for receiving electrical impulses, short) and multiple signal-sending outgrowths (axons, single projection for sending electrical impulses, long and myelinated for insulation) which end in a terminal button.
These interact through the synapse with dendrites on other neurons. These characteristics are shown in Figure \(\PageIndex{1}\).
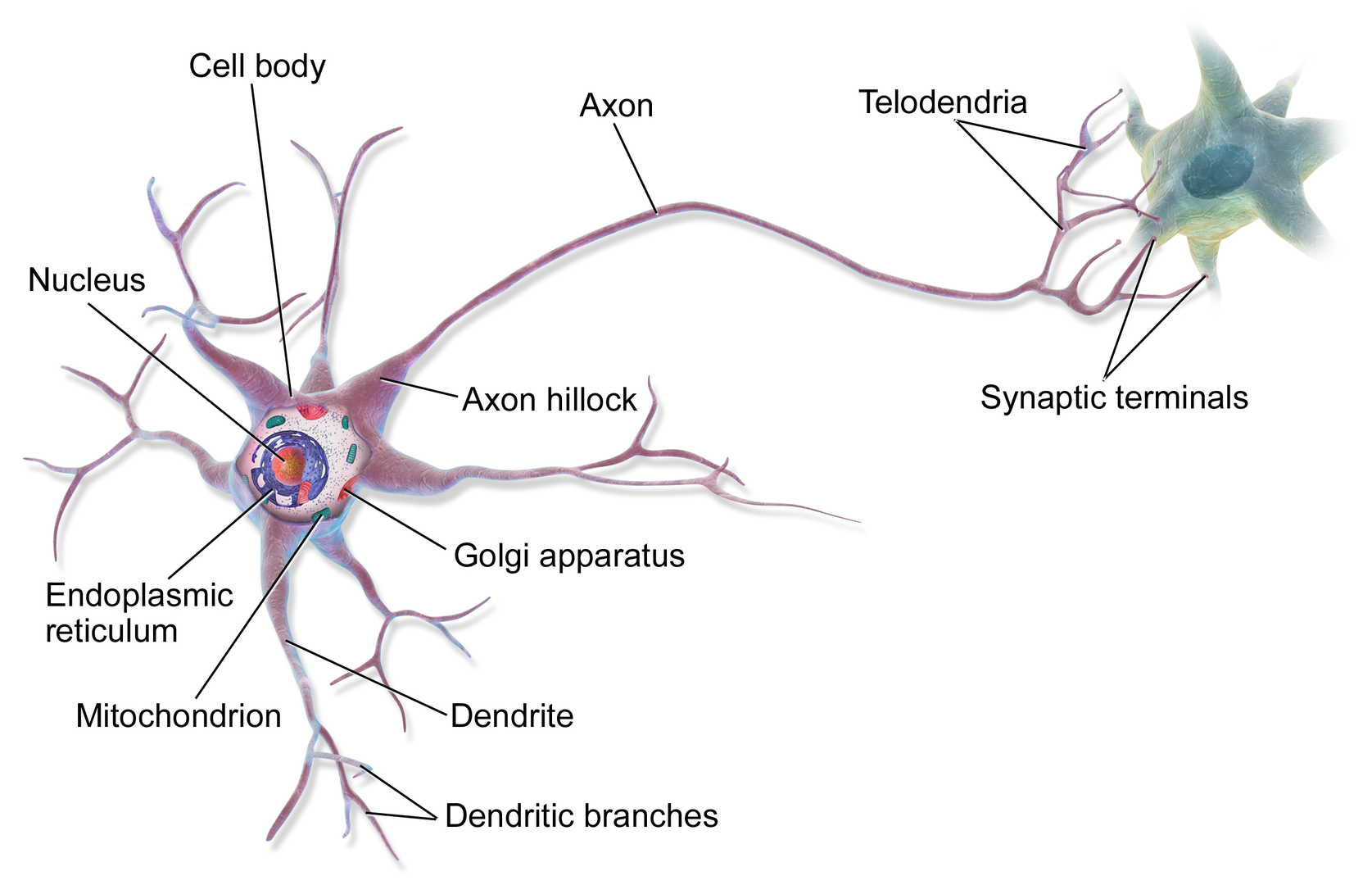
The high-resolution map of the human cortex shows that an axon most commonly (96%) forms one synapse with a target cell. 3% of the axons form 2 synapses with the target cell. However, neurons that had lots of input onto their dendrites (they were highly innervated with more than 3000 synapses to their dendrites) could form 7 or more synapses from their axon to a target cell.
A presynaptic neuron can stimulate an adjacent postsynaptic neuron by releasing a neurotransmitter into the synapse between the cells, which binds to a receptor in the membrane of the postsynaptic cell, stimulating the cell, as shown in Figure \(\PageIndex{2}\).
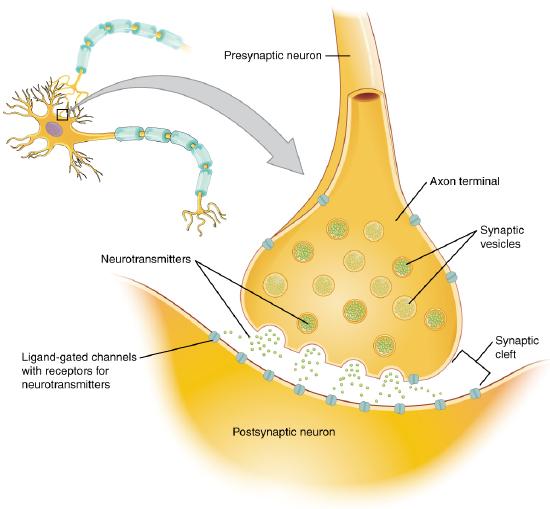
Figure \(\PageIndex{2}\): The synapse. https://cnx.org/contents/FPtK1zmh@8.25:fEI3C8Ot@10/Preface..Creative Commons Attribution 4.0 International license.
We will discuss the events that cause the post-synaptic cell to "fire," but we will not discuss the immediate events that lead to the release of neurotransmitters by the presynaptic neuron.
Neurons (like all cells) have a transmembrane potential across the membrane. Transient changes in the membrane potential are associated with neuron activation or inhibition. This arises partly due to the imbalance of ions across the membrane, which the Na+-K+-ATPase establishes in the membrane. This electrogenic antiporter transfers 3 Na ions out of the cytoplasm for every 2 K ions it transports in. Likewise, Cl- has a much higher level outside the cell. Membrane potentials are determined by the size of the ion gradients across the membrane and by the differential permeability of membranes to ions. As we saw previously, synthetic bilayer membranes are not very permeable to ions, as shown in Table \(\PageIndex{1}\) below.
| ION | PERMEABILITY (cm/s) |
|---|---|
| sodium | <1.6 x 10-13 (lowest) |
| potassium | <9 x 10-13 |
| chloride | <1.5 x 10-11 (highest) |
Table \(\PageIndex{1}\): Ion permeability of phosphatidyl serine vesicles
Sodium would be expected to have a lower permeability than potassium since it has a higher charge density. It is also the largest in effective size due to the larger hydration sphere around the ion arising from its higher charge density. Chloride would be expected to have the highest permeability since it has the lowest charge density (due to the repulsion of the electron cloud in the negative ion). Intracellular charged proteins (which are mostly negative) are not permeable and help create the negative charge imbalance across the membrane.
Much work has been done on the giant axon of the squid, which has uniquely high intracellular potassium (400 mM compared to 20 mM outside) and high extracellular fluid sodium (440 mM vs 50 mM inside) and chloride (560 mM vs 52 mM inside). Mammalian cell concentrations are much lower, but the relative size of the gradient is about the same. Typical ion concentrations and permeabilities for mammalian membranes are shown in Table \(\PageIndex{2}\) below.
| Ion | Cell (mM) | Blood (mM) | Permeability (cm/s) |
|---|---|---|---|
| potassium | 140 | 5 | 5 x 10-7 |
| sodium | 5-15 | 145 | 5 x 10-9 |
| chloride | 4 | 110 | 1 x 10-8 |
| X- (negative macromolecules) | 138 | 9 | 0 |
Table \(\PageIndex{2}\): Typical ion concentrations and permeabilities for mammalian membranes
How can we account for the markedly greater permeabilities of ions (1000x to 1,000,000x) in mammalian cell membranes compared to synthetic lipid vesicles? Previously, we showed that glucose has a greater permeability through red blood cell membranes than synthetic liposomes because of a membrane receptor allowing facilitated diffusion across the membrane and down a concentration gradient. The same thing is true of ion permeabilities in intact biological membranes. These membranes have different selective ion channels (non-gated - always open, and gated - open only after specific conformational changes). The non-gated channels dramatically increase the permeability of membranes to ions, as the glucose transport protein increases the permeability to glucose. It turns out that this differential permeability contributes to the transmembrane potential. Ion channels in nerves and muscles can move ions across the membrane at a rate of up to 109/s, comparable to kcat for the best enzymes.
If we envision channels as pores, how can we account for the channel's selectivity to specific ions? A larger pore should admit any ion less than a maximal size for the pore, so it is hard to imagine the nature of the selectivity filter. Because of this, many people discounted the ideas of channels in favor of a transporter, which would bind the ion selectivity and then, through conformational changes, move the ion across (much like the Na/K ATPase we discussed in the previous guide). This model could not, however, account for the incredible rates of ion flux across the membrane. Selectivity can be accounted for by a channel that contains a narrow opening that acts as an ion sieve. The ion loses most of its hydration sphere and forms specific interactions with amino acid side chains in the pore region. Such an interaction would be transient and not too tight since the ion must pass through the membrane. As we will see later, these ion channels:
- pass ions down a concentration gradient in a thermodynamically favorable process
- are specific for certain ions (although a few are less selective and will pass Na, K, Ca, and Mg ions)
- allow ion flow through either ungated or gated channels.,
- saturate with increasing ion concentration (even though concentration increases, the ions have a greater thermodynamic drive to pass through the channel). This is consistent with the ion binding at a selectivity filter in the narrow part of the pore. The KD for the interaction is usually in the mM range and indicates weak binding with large dissociation rate constants (koff).
Transmembrane Potentials
Several questions arise about ions' distribution and the magnitude of the transmembrane potential.
- How are the ion gradients established?
- How does the transmembrane ion distribution contribute to the membrane potential?
- How can the resting electrochemical potential and the ion distribution be maintained?
The answer to these questions will be illustrated using studies on two types of brain cells, glial cells (which function as protectors, scavengers, and feeders for brain neurons) and neurons. Both types of cells have transmembrane potentials.
Glial Cells
Different mechanisms can establish the transmembrane ion gradients for ions. One uses ion-specific ATPases (P-type ion transporters), as we discussed with the Na/K ATPase. This transporter ejects three sodium ions from inside the cell for every two potassium ions it transports in, all against a concentration gradient. Since it is an electrogenic antiporter, it helps generate the potential. Specific ion channels also contribute (as described below) to the transmembrane gradients and potentials.
The harder question is how the ion distribution contributes to the membrane potential. Two things must occur for a membrane potential to exist: First, there must be a concentration gradient of charged ions (for example, sodium, potassium, or chloride) across the membrane. Second, the membrane must be differentially permeable to different ions. If the membrane were entirely impermeable to ions, then no movement of ions across the membrane could occur, and no membrane potential would arise. If membranes are differentially permeable to the ions, an electrical potential across the membrane can arise. Remember, synthetic bilayers are quite impermeable to ions, given the hydrophobicity of the internal part of the bilayer. Likewise, it is quite impermeable to glucose. It turns out that glial cells appear to have only a non-gated potassium channel, which allows the outward flow of potassium ions down the concentration gradient. The inside will then have a net negative charge since impermeable anions remain.
The chemical potential gradient causes this outward flow of potassium ions. As more ions leave, the inside gets more negative, and a transmembrane potential develops, which resists further efflux of potassium. Eventually, they balance, and the net efflux of potassium stops. The resting transmembrane potential reaches around -75 mV, the value obtained from the equations we will derive below. Since glial cells appear only to express a non-gated (or leakage) potassium channel, their resting potential equals the potassium equilibrium potential. Figure \(\PageIndex{3}\) shows hypothetical transmembrane potentials (Ψ), and the electrical and chemical potentials (ΔG) for K+ loaded vesicles with and without a non-gated K+ channel

As shown previously, very little K+ efflux is required to develop a transmembrane potential. At equilibrium, the K+IN is simply shown as < 0.1 M while K+OUT is shown as > 0.1 M
A PhET simulation shows the differences between non-gated (leakage, always open) and gated channels (opened in response to stimuli like ligands, transmembrane potential, mechanical forces, etc).
Neurotransmitter Activation of Neurons
What happens when a neurotransmitter binds to a receptor on the postsynaptic cell? We will study two examples. The first is the simplest: binding the neurotransmitter acetylcholine, released by a motor neuron, to its receptor on the muscle. This region is called the neuromuscular junction. Acetylcholine binding will lead to a transient depolarization of the muscle cell. Next, we will discuss the interaction of a neurotransmitter with a postsynaptic neuron in the central nervous system. This is a much more complex system. Their differences are described below:
In neurons interacting with muscles:
- Most muscle fibers are innervated by only one neuron, a motor neuron
- Neurotransmitter release at the neuromuscular junction leads only to muscle excitation, not inhibition.
- All fibers are excited by the same neurotransmitter, acetylcholine.
In the central nervous system, life is more complicated:
- Stimuli are received from hundreds to thousands of different neurons.
- Nerves receive both excitatory and inhibitory stimuli from neurotransmitters.
- Different kinds of receptors are present to receive stimuli, which control the activity of different channels.
- Neuronal ion channels are gated by various mechanisms in addition to changes in membrane potential, including gating by heat, cold, stretch, or covalent modification.
- Most nerve cells have a resting potential of about -65 mV compared to -90 mV for a muscle cell.
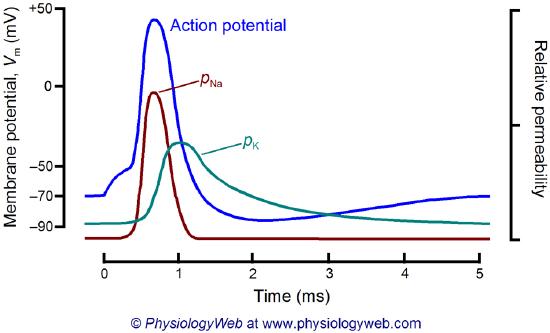
What happens when a neurotransmitter binds to the receptor on the postsynaptic cell? A depolarization occurs (mediated by conformational changes in the transmitter-receptor complex), raising the membrane potential from the resting equilibrium level. What happens next depends on the identity of the post-synaptic cell. In the muscle cell, the rising potential caused by acetylcholine binding ultimately leads to muscle contraction by opening the intracellular organelle membrane calcium channels. In a neuron, the rising potential triggers an action potential by opening voltage-gated sodium channels. The potential rises to about + 35 mV but does not reach the Na+ ion equilibrium potential, because the high positive potential opens a voltage-gated potassium channel. The potential then falls until it reaches the K+ ion equilibrium potential when the cells are hyperpolarized. It slowly then relaxes back to the resting potential of -60 mV. This wave of changes in potential sweeps down the post-synaptic cell membrane and is the basis for the "firing" of the neuron. A plot of transmembrane voltage changes vs time for a typical action potential is shown in Figure \(\PageIndex{4}\).

Figure \(\PageIndex{5}\) shows the actual changes in ion permeability in various phases of the action potential (replace figure, seek permission)
mine

Figure \(\PageIndex{5}\): Na+ and K+ permeability during the action potential
Figure \(\PageIndex{6}\) shows an animation of a neuron firing, showing all the key players. (produced by PhET, University of Colorado, Boulder).
Proteins of the Neural Synapse
We must now account for the rise and fall of the membrane potential to a variety of neurotransmitters, including the cholinergic transmitters (ex., acetylcholine), catecholamines (dopamine, epinephrine, norepinephrine), amino acid derivatives (ex., Glu, Asp, N-methyl-D-Asp, Gly, gamma-amino-butyric acid -GABA), and peptides (endorphins, enkephalins). We will consider five membrane proteins as shown below in Figure \(\PageIndex{7}\).

We have explored many of these in detail in Chapter 11.3: Diffusion Across a Membrane—Channels, so in the following section, we will focus on just one new one, the chloride channel.
Na+-K+-ATPase: It transports sodium and potassium ions against a concentration gradient using ATP as an energy source. The protein is a sodium-dependent ATPase. The membrane potential could not be maintained without this protein since the sodium and potassium gradient would collapse. It also contributes to the potential since it is an electrogenic antiporter. (We have also seen that ungated potassium and sodium channels are present.)
Neurotransmitter receptor: The receptors we will consider here are typically ligand-gated ion channels. Once the ligand binds, a conformational change occurs in the protein, allowing a flow of ions down a concentration gradient. Depending on the nature of the ion, the channel either initiates depolarization (when Na+ enters from the outside and raises ΔΨ) or inhibits depolarization (when Cl- enters from the outside and lowers ΔΨ). When chloride channels open, they hyperpolarize the transmembrane potential. Stimulatory neurotransmitters (like glutamate) lead to depolarization of the membrane, while inhibitory neurotransmitters (like gamma-aminobutyric acid) lead to hyperpolarization of the membrane (making the potential more negative).
Na+ channel (voltage-gated): When the ligand-gated channel depolarizes the membrane to some threshold value, sodium channels undergo a conformational change and open, allowing Na+ ions to flood into the cell, raising the potential to a positive value. 33 mV (a value lower than the equilibrium sodium potential). This membrane protein is a voltage-gated channel, not a ligand-gated one.
Two potent neurotoxins, tetrodotoxin (from Pufferfish) and saxitoxin, bind to the channel and act as antagonists (inhibit the activity of the receptor by blocking sodium influx). Their structures are shown in Figure \(\PageIndex{8}\).

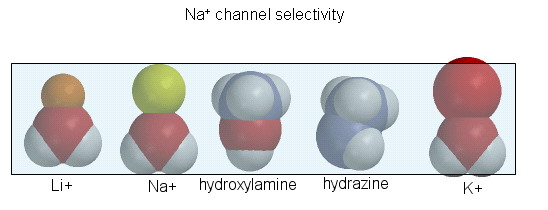
The guanidino group of the tetrodotoxin appears to bind with high affinity to the channel's entrance that interacts with the hydrated sodium ion. Affinity chromatography using tetrodotoxin beads has been used to purify the protein. Figure \(\PageIndex{9}\) shows the relative sizes of ions used to probe size requirements for the channel. The hydrated K+ ion can not pass through.
Depolarization of the membrane potential may result in an outward movement and rotation of the positively charged helices containing Lys and Arg side chains, presumably forming salt bridges (ion-ion interactions) with negatively charged side chains within the protein. Depolarization of the membrane breaks a few salt bridges and effectively leads to the movement of 1 or 2 charges on the helix through the membrane. Work occurs when charges are moved through an electric field. Work is also related to the ΔG for the system, which is also dependent on the ratio of the open and closed forms of the channel. Other voltage-gated ion channels (for potassium and chloride) have a similar membrane topology and an S4 voltage-sensor helix.
K+ channel (voltage-gated): When the membrane potential reaches around +25 mV or so, the K+ channel, a voltage-gated membrane protein, alters its conformation, allowing K+ efflux from the cells. This lowers the potential until it reaches the potassium equilibrium potential. It then slowly relaxes back to the cell resting potential of about -60 mV.
Cl- channel: If these channels (typically ligand-gated) are open, they will hyperpolarize the cell and make it more difficult to fire.
The selectivity filter comprises many stacked rings of oxygens that can interact with a dehydrated K ion but not with a dehydrated Na+ ion, which can not approach close enough to form significant interactions. Surrounding the filter are twelve aromatic amino acids that constrain the size of the pore opening. The interactions of the filter O's with the K ion make up for the energetically disfavored dehydration of the ion. The filter contains two K ions, which repel each other, assisting in the vectorial discharge of the ions through the membrane. These ions must form weak interactions with the selectivity filter. The actual pore is hydrophobic primarily, which facilitates the flow-through of the ions. The central cavity of the pore can hold some water molecules in addition to the K ions, which helps stabilize the ion in the middle of the pore.
Inhibitory Neurotransmitters - The GABA receptor
The main inhibitory neurotransmitters are GABA (gamma-aminobutyric acid), made from glutamic acid through decarboxylation of the α-C group, and glycine. They bind to transmitter-gated chloride channels, which, when open, hyperpolarize (make more negative) the transmembrane potential. Benzodiazepines (like Valium and Librium - anti-anxiety and muscle-relaxing agents) and barbiturates (like phenobarbital hypnotics) bind at allosteric sites on the GABA receptor and potentiate the binding of each other and GABA. This receptor is also affected by alcohol and anesthetics. Let's focus on the GABAA channels (also called the GABA(A) receptor or GABA(A)R) since they bind so many interesting pharmaceutical drugs.
Figure \(\PageIndex{10}\) shows an interactive iCn3D model of the human full-length heteromeric α1-β3-γ2L GABA(A)R in complex with picrotoxin, GABA, and megabody Mb38 (6huj) derived from cryo-EM (loads slowly).
R_-picrotoxin__GABA_(6huj).png?revision=2&size=bestfit&width=378&height=338)
This structure is a Type-A γ-aminobutyric (GABA A ) receptor. It consists of two alpha chains (orange), two beta chains (magenta), and one gamma chain (brown). Two GABAs (spacefill, CPK colors) are bound between the orange and magenta subunits (between the α and β chains). Picrotoxin is shown in spacefill CPK colors in the central pore (bottom), near the cytoplasmic end (blue bilayer dummy atoms). The extracellular domain (above the red) has glycans shown in SNFG cartoon form and shown in Figure \(\PageIndex{11}\).

The structure of the receptor bound to many pharmacological agents has been solved. The structure of GABA (the agonist), bicuculline (a competitive antagonist), the benzodiazepines alprazolam (Valium) and diazepam (Xanax), a channel blocker (picrotoxin), ethanol, and Ro-15-4513 (an ethanol antagonist) are shown in Figure \(\PageIndex{12}\).

(\PageIndex{12}\): Structures of GABA-R binding molecules -GABA (the agonist), bicuculline (a competitive antagonist), the benzodiazepines alprazolam (Valium) and diazepam (Xanax), a channel blocker (picrotoxin), ethanol, and Ro-15-4513 (an ethanol antagonist)
The benzodiazepine diazepam (Xanax) binds at an allosteric site and promotes GABA binding, so it is considered an "indirect agonist”. It is also an anxiolytic (a drug used to reduce anxiety) and an anticonvulsant. Ethanol activates the inhibitory GABA channel. Too much ethanol leads to hyperpolarized cells, so neural responses are greatly inhibited, possibly leading to death. Ethanol acts synergistically with benzodiazepines, which makes this combination so lethal.
Figure \(\PageIndex{13}\) shows an interactive iCn3D model of the human full-length heteromeric α1-β3-γ2L GABA(A)R in complex with diazepam (Valium), GABA (6HUP) (loads slowly). https://structure.ncbi.nlm.nih.gov/i...fTZMqePnpdwTy8
R_in_complex_with_diazepam_(Valium)_and_GABA_(6HUP).png?revision=1&size=bestfit&width=384&height=394)
Figure \(\PageIndex{13}\): Human full-length heteromeric α1-β3-γ2L GABA(A)R in complex with diazepam (Valium), GABA (6HUP). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...fTZMqePnpdwTy8
The structure shows two alpha chains (orange), two beta chains (magenta), and one gamma chain (gray). Two GABAs (spacefill, CPK colors) are bound between the orange and magenta subunits (between the α and β chains). Three Valiums (spacefill, CPK) are shown in the structure. One is bound to an allosteric site between the orange (alpha) and gray (gamma) chains. Two others are bound within the bilayer, this time between the alpha (orange) and beta chains (magenta). Different benzodiazepines appear to bind in slightly different sites, and binding elicits their specific effects.
A drug, Ro-15-4513, was developed in the 1980s that antagonizes the effect of ethanol. It has a complicated pharmacology. It is considered a benzodiazepine “partial inverse agonist”. It has no effect by itself. It reverses the anticonvulsant effects of benzodiazepines and blocks the Cl- effects of ethanol, so it is an inverse agonist of GABA-mediated Cl- flux. Hence, it antagonizes the effect of benzodiazepines and alcohol. If given to intoxicated mice, they act normally! Such drugs have not reached the market as their use poses significant ethical issues.
The effect of pharmacological agents on the GABA receptor
In the “voltmeters” below, draw an arrow indicating if the transmembrane potential becomes more negative or more positive for the conditions given.

- Answer
-
Add texts here. Do not delete this text first.
Metabotropic Neural Receptors
Some signaling molecules, whose effects are regulated by kinases (β-adrenergic and some olfactory signals by PKA, and acetylcholine by PKC, for example), are neurotransmitters. In all the previous examples, neurotransmitters directly gate inactive ion channels. These types of membrane receptors are classified as ionotropic. Typical examples of neurotransmitter-gated ion channels are the acetylcholine receptor in neuromuscular junctions and the central nervous system's Glu, Gly, and GABA receptors. These receptors are multimeric proteins. Receptors with direct gating of ion flow are fast, with activities that last milliseconds, and are used in eliciting behavioral responses.
However, ion channels can also be gated indirectly when the neurotransmitter binds to its receptor and leads to downstream events, subsequently opening an ion channel distinct from the receptor. In this case, the occupied receptor indirectly communicates to an ion channel through a G protein, for example. Examples of this indirect gating of ion channels include the serotonin, adrenergic, and dopamine receptors in the brain. These receptors are classic single-protein serpentine GPR receptors with seven transmembrane helices and intracellular domains that can interact with G proteins as described above to increase second messenger levels (cAMP, DAG) in the neuron. The receptors are classified as metabotropic since they must activate a series of metabolic steps before ion channels are opened. The second messengers can either activate kinases in the cell, which phosphorylate ion channels to open or close them, or bind directly to the channel and modulate its activity through an allosteric conformational change. In some cases, the G protein directly acts on the ion channel. These different ways are described in Figure \(\PageIndex{14}\).

In contrast to direct gating, receptors that indirectly gate ion channels have slow activities that last seconds to minutes. These receptors are usually involved in modulating behavior by changing the excitability of neurons and the strength of neural connections, hence modulating learning and memory. These changes can occur in many ways, summarized below:
Phosphorylating ion channels: Receptors that act through a second messenger system can change ion channel activity by activating kinases that phosphorylate the channels. This may:
- open the channel normally closed at the resting potential, producing an effect like gating.
- Close a channel that usually opens at the resting potential (such as non-gated K channels, which, when closed, would depolarize the cell and make it more excitable).
Gα interaction with ion channels:
- the Ga subunit of the G protein interacts with K channels after stimulation of the CNS Acetylcholine receptor, opening the channel and hyperpolarizing the cell
Second messenger interaction with ion channels:
- cGMP opens cation channels in retinal cells after activation of the photoreceptor by photons
- cAMP opens cation channels in olfactory cells after activation of the olfactory receptor by odorants.
Second messenger effects on proteins other than ion channels (usually different receptors):
- the β-adrenergic receptors are phosphorylated by PKA and PKC (activated by stimulation of a different neurotransmitter receptor linked through a G protein to produce increased levels of second messengers cAMP and diacylglycerol). When phosphorylated, the β-adrenergic receptor, itself activated through G protein) can't bind Gs. This attenuates the response of the β-adrenergic receptor to its neurotransmitter, which leads to desensitization to that signal.
Second messengers regulate gene expression:
- cAMP-activated PKA can phosphorylate an inactive transcription factor in the cell, which then can bind to a section of DNA called the cAMP Response Element (CRE), which is upstream of specific genes, leading to the transcription of the genes. The transcription factor is called CREB for cAMP Response Element Binding protein. Example: Tyrosine hydroxylase (a monooxygenase) is a key enzyme in synthesizing epinephrine and norepinephrine. The activity of this protein is increased when PKA phosphorylates it. Hence, this modification of the already present protein can increase its activity quickly. If an animal is subjected to severe or long-term stress (cold or immobilization), presynaptic cells with norepinephrine will be stimulated to release the neurotransmitter. This requires continual synthesis of the neurotransmitter by the presynaptic cell. The increase in the synthesis of this neurotransmitter is caused by the presynaptic cell being stimulated by another neuron, which leads to increased levels of cAMP, and ultimately activation of CREB, which increases transcription of the hydroxylase gene.
Caffeine
Caffeine produces a state of arousal in the central nervous system. High levels block the binding of an inhibitory neurotransmitter, adenosine, to the A2A adenosine receptor. Without caffeine, adenosine levels rise during the day, promoting interaction with its receptor, leading to increased sleepiness and lack of concentration. When adenosine binds normally to its receptors, it activates the adenylate cyclase cascade, which activates PKA, leading to changes in the phosphorylation state of many proteins inside the cell, including protein phosphatase 2A. These changes inhibit neural firing. Caffeine blocks these changes.
Hallucinogenic drugs
Illicit drugs like LSD, psilocybin, and ecstasy can produce hallucinations as they have profound effects on consciousness and perception of self and reality. Recent clinical studies have shown that under tightly controlled conditions and doses, these drugs might have significant therapeutic effects in the treatment of mental health issues such as depression and post-traumatic stress disorder. Hence, their mechanisms of action have been the source of many studies.
All these drugs bind to the human serotonin 2A receptor (5-HT2AR), a metabotropic GPCR receptor of serotonin (5-hydroxytryptamine). When serotonin binds, it leads to GPCR signaling, partly through beta-arrestins. These adapter proteins form complexes with ligand-bound and activated GPCRs and GPCR protein kinases.
Structures show that LSD binds to the orthosteric binding site (i.e., the active site, not an allosteric site) for serotonin. The study also found that serotonin and psilocin binding extends into an adjacent site called the extended binding pocket (EBP). These differences suggest that it should be possible to design agonists that have therapeutic, but not hallucinogenic, properties. Serotonin binding does not cause hallucinations. A drug, IHCH-7086, in mouse studies does not seem to provoke hallucinations but appears to have antidepressant effects (based on observations of mouse behaviors like twitching and freezing). The structures of serotonin (5HT) and psychotropic drugs that bind the 5HT2AR are shown in Figure \(\PageIndex{15}\).

Figure \(\PageIndex{15}\): Structures of serotonin (5HT) and psychotropic drugs that bind the 5HT2AR
Figure \(\PageIndex{16}\) shows a model of the serotonin receptor (5HT-2A) with each of the bound drugs. Note the different occupancy of the orthosteric and extended binding pockets.
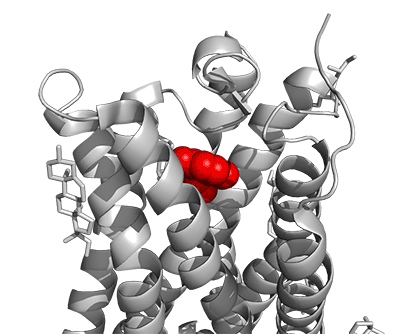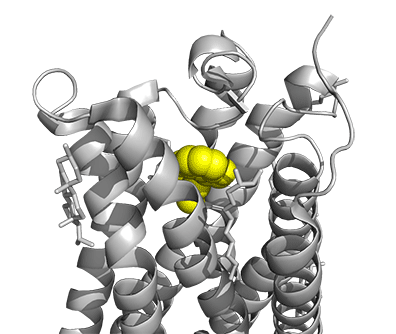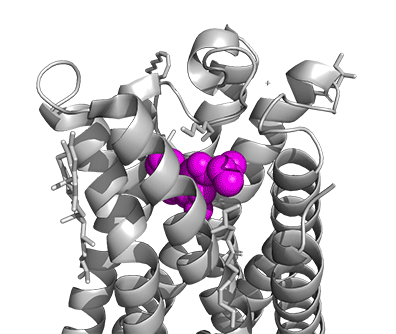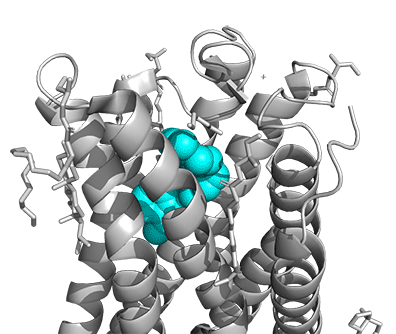
The GPCRs have been aligned for each of the poses. The ligands are represented in spacefill in a single color, as listed below:
- red: serotonin (5HT), the physiological agonist, 7WC4
- yellow: psilocin, 7WC5 LSD, 7WC6
- magenta: LSD, 7WC6
- cyan: IHCH-7086
An alternative explanation for why serotonin does not cause hallucinations and why the other agonists do is that, in addition to binding plasma membrane GPCRs, the agonist might have additional effects that are associated with hallucinations by binding intracellular membrane GPCRs.
The cerebral cortex plays a key role in what it means to be human. It is key to higher-level processing involving thought, learning, emotion, personality, language, and memory. As such, its dysregulation is involved in mental health conditions such as depression, et al., in which synaptic plasticity is decreased as evidenced by a reduction in dendritic structure required for synaptic connections. The serotonin (5HT) reuptake inhibitors (SSRIs) used to treat depression appear to increase neural plasticity and connections over time (so they don't give immediate responses). Hallucinogens that target the 5-HT2A receptor (5-HT2AR) also promote neuroplasticity. However, neuron growth in cortical cultures is not affected by serotonin. What is the mechanism that allows various combinations of neuron plasticity and hallucinations, all from the same receptor?
One factor determining these differential effects is that serotonin (5-HT) is more polar and cannot readily enter cells. At the same time, the HT2AR agonists that are hallucinogenic are less polar (more lipophilic) and could potentially enter cells where they might elicit hallucinogenic effects. Intracellular hallucinogens would bind internal 5-HT2AR receptors, which are found in cortical neurons on the Golgi and other organelles that are more acidic than the cytoplasm or extracellular environment. (In fact, in cortical neurons, the main location of the 5-HT2ARs is intracellular.) This might lead to more prolonged retention of intracellular hallucinogens and longer signaling (LSD has a profound hallucinogenic effect that lasts for 10 hours or more), leading to neuronal growth as well.
If serotonin is given to wild-type mice, no hallucinations occur, as evidenced by the lack of a head twitch response (HTR). However, if given to mutant mice who expressed serotonin transporter (SERT) on cortical neurons, the HTR response was observed. The import of serotonin led to neuroplasticity. It could be that serotonin is not the physiological ligand for intracellular HT2ARs. It might also imply that, as with the case of endocannabinoids, we have endogenous psychedelics. The subtle difference in the binding of serotonin and the hallucinogens in Figure 16 might have little to do with their tendency to produce hallucinations.
Recent Updates:
Much is still not known about how antidepressants work and what causes delays in their therapeutic effects. Increased synaptic connections through expanded neuroplasticity appear to be required for their therapeutic effects. The major action of drugs like Prozac, a serotonin reuptake inhibitor, is blocking the serotonin transporter (SERT), increasing extracellular serotonin levels in the neural synapse. Tricyclic anti-depressants as well as monoamine oxidase inMAOIstors (MAOI), also increase monoamine neurotransmitters in the synapse with delayed therapeutic effects (the monoamines act quickly, however). Perhaps these agents bind other receptors!
They do. The binding of both typical and fast antidepressants also occurs in the transmembrane domain of tyrosine kinase receptor 2 (TRKB), the brain-derived neurotrophic factor (BDNF) receptor, which is linked to increases in neuroplasticity. The receptor also binds cholesterol, which modulates its activity. The antidepressant binding site is formed on dimerization of their transmembrane domains. Mutations in the transmembrane region block the efficacy of the antidepressants.
LSD and other psychedelics also produce fast and long-lasting antidepressant effects promoted by increases in neuroplasticity. Studies have shown that LSD and psilocin bind to slightly overlapping sites in the transmembrane domain of the BDNF receptor as antidepressants, but with a 1000-fold higher affinity. If the LSD binding site on the serotonin 2A receptor (5-HT2A) is blocked, LSD still has antidepressant and increased neuroplasticity effects.
Figure \(\PageIndex{17}\) shows the interaction of LSD and the metabolite of psilocybin, psilocin (PSI), with the TRKB receptor.

Figure \(\PageIndex{17}\): Characterization of the psychedelics binding site in the TrkB TMD. Moliner, R., Girych, M., Brunello, C.A. et al. Psychedelics promote plasticity by directly binding to the BDNF receptor TrkB. Nat Neurosci 26, 1032–1041 (2023). https://doi.org/10.1038/s41593-023-01316-5. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panel a–c show representative MD snapshots showing the binding pocket for LSD (purple) (a) and PSI (green) (c) in the extracellular-facing crevice of the TrkB TMD dimer (gray). Side chains (yellow) of relevant binding site residues are displayed. A structural model of full-length TrkB dimer (gray) embedded in a lipid membrane is shown with bound BDNF (blue) and LSD (purple) (b).
Panel d shows in silico binding free energy estimations for fluoxetine, LSD, and PSI. Each free energy estimate (ΔG, circles) and its statistical error (SE, bars) were estimated from a separate set of FEP simulations (n = 1). Dissociation constants are given as a range with upper and lower bounds converted from ΔG − SE and ΔG + SE, respectively.
Panels e,f show chemical structures of LSD (e) and PSI (f) with atom numbers annotated.
Panel i shows the distributions of TMD dimer C-terminal distance and that LSD and PSI stabilize the cross-shaped conformation of TrkB favorable for receptor activation in a 40 mol% CHOL membrane. Lines represent the mean distribution, and bands represent the standard errors (n = 10 independent simulations). TMD conformations corresponding to indicated C-terminal distances and drug-bound states are shown in the inset.
Figure \(\PageIndex{18}\) shows the different binding modes of LSD and Fluoxetine (Prozac), a selective serotonin reuptake inhibitor (SSRI) for TrkB. Fluoxetine is used to treat depression, obsessive-compulsive disorder (OCD), bulimia nervosa, and panic disorder.

Figure \(\PageIndex{18}\): Different TrkB binding modes of LSD and fluoxetine. Moliner et al., ibid.
Panels a,b, show representative snapshots of atomistic MD simulations showing the front (a) and back (b) views of the binding pockets for LSD (purple) and fluoxetine (yellow) in the extracellular-facing crevice of TrkB TMD dimers. Side chains of relevant binding site residues are displayed. Superimposed structures of TrkB optimally bound to LSD or fluoxetine reveal that, while some residues involved in binding are shared (Y433 and V437), the binding modes are different. Fluoxetine binds at a site deeper within the dimer, locking the TMD dimers in a more open cross-shaped conformation (distance between the center of mass L451–L453 Cα atoms of each monomer ~20 Å). In contrast, LSD binds closer to the N-terminus of the TrkB TMD and establishes more stable interactions with the dimer: a hydrogen bond between the oxygen atom of the diethylamide group of LSD and the Y433 residue of one monomer, and pi-stacking of the aromatic backbone of the drug with the Y433 residue of the second monomer, locking the TMD dimer in a tighter cross-shaped conformation (L451–L453 Cα distance ~17 Å) compared with fluoxetine. Drugs are shown in van der Waals representation. Oxygen, nitrogen, and hydrogen atoms are shown in red, blue, and white, respectively.
Summary
In this chapter, we explore the molecular and biophysical foundations of neural signaling, focusing on how ions traverse cellular membranes via specialized ion channels and how neurotransmitters modulate these processes to initiate and regulate electrical activity in neurons. Key topics include:
-
Ion Channels and Membrane Permeability:
Ion channels are integral membrane proteins that provide pathways for the rapid, selective movement of ions (e.g., Na⁺, K⁺, Cl⁻) across the otherwise impermeable lipid bilayer. The chapter contrasts synthetic lipid bilayers—which have extremely low ion permeability—with biological membranes that express various gated channels and transporters. These channels are responsible for the establishment and dynamic modulation of the transmembrane potential. -
Generation and Maintenance of Membrane Potentials:
The resting membrane potential arises from a combination of ion gradients—maintained by pumps such as the Na⁺/K⁺-ATPase—and differential membrane permeability. Glial cells, for example, maintain a resting potential largely defined by a non-gated potassium leakage channel. This potential is crucial for neuronal excitability and signal propagation. -
Action Potential Dynamics:
Neuronal signaling involves the generation of action potentials, rapid fluctuations in membrane potential caused by the sequential opening and closing of voltage-gated sodium and potassium channels. The chapter discusses the phases of depolarization and repolarization, and how ion fluxes contribute to the rapid firing of neurons. -
Synaptic Transmission and Neurotransmitter Receptors:
At synapses, presynaptic neurons release neurotransmitters that bind to receptors on the postsynaptic cell. Ionotropic receptors, such as the acetylcholine receptor at the neuromuscular junction, directly gate ion channels to produce fast, transient responses. In contrast, metabotropic receptors, which are typically G protein-coupled, trigger intracellular signaling cascades that modulate ion channel activity and gene expression, producing slower and longer-lasting effects. -
Ion Channel Selectivity and Neurotoxins:
The selectivity of ion channels is achieved through specialized narrow pores that strip ions of part of their hydration shells and coordinate them via specific interactions. Neurotoxins like tetrodotoxin and saxitoxin, which block sodium channels, provide insights into channel structure and function and underscore the importance of precise ion flow in neural activity. -
Integration of Neural Signaling:
Neural communication is not only determined by the rapid opening and closing of channels but also by the coordinated activity of multiple receptor types and signaling proteins. This integration underpins complex processes such as synaptic plasticity, learning, memory, and behavior. The chapter highlights how disruptions in these mechanisms can contribute to neurological disorders.
Overall, the chapter presents a detailed view of how ion channel function, neurotransmitter receptor dynamics, and synaptic architecture converge to produce the electrical signals that underpin neural activity. By linking structural features to functional outcomes, it sets the stage for understanding the complex interplay between molecular events and higher-order brain functions in both health and disease.