
25.2: RNA Processing
- Page ID
- 15198


Post-transcriptional modifications of rRNA and tRNA will be topics of Chapter 27 as their structure and function in protein synthesis will be a focal point. Thus, this section will focus on post-transcriptional modifications of mRNA. We'll spend most of our time on eukaryotic RNA processing.
Prokaryotic RNA Processing
First, let's take a brief look at a fascinating feature of transcription in bacterial cells. Bacterial cells do not have extensive post-transcriptional modifications of mRNA primarily because transcription and translation are coupled processes. Bacterial cells lack the physical barrier of a nucleus, which allows transcription and translation machinery to function at the same time, enabling the concurrent translation of an mRNA while it is being transcribed (Fig 26.2.1). As mRNA is synthesized in prokaryotes, a ribosome binding motif called the Shine-Dalgarno sequence, located in the 5' untranslated region of the mRNA emerges early, allowing the ribosome to bind and translation to occur. In addition, the protein N-utilzation Substance, better known as NusG, plays a critical role. NusG has three separate domains and the functions of two of them are known. The NusG N-terminal domain (NusG-NTD) can bind to RNAP, whereas the C-terminal domain (NusG-CTD) can combine with the NusE (RpsJ) component of ribosomes. These two functions of NusG enable transcription to be coupled with translation. NusG CTD can also bind to Rho to terminate transcription, as shown in Figure \(\PageIndex{1}\).
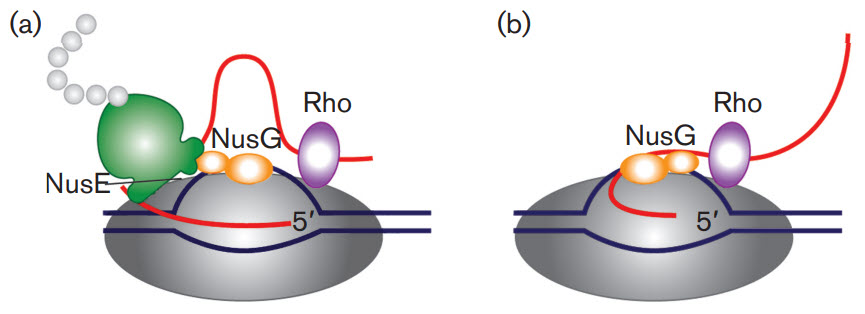
Figure \(\PageIndex{1}\): The roles of NusG in transcription/translation coupling. (a) Composition of an active RNAP complex. RNAP is shown in dark grey, DNA in blue and nascent RNA in red. The ribosome is shown in green with the nascent polypeptide chain in light grey; the bulge in the small subunit denotes the location of NusE (RpsJ). NusG is shown in orange: its shape denotes two functional sections. The larger section denotes the N-terminal domain, which binds to RNAP. The smaller section denotes the C-terminal domain, which interacts with NusE in situ. Rho is shown in purple. (b) After the translation is completed, NusG remains bound to RNAP and may also bind to Rho through the C-terminal domain leading to the termination of transcription. Figure from: Cortes, T., and Cox, R.A. (2015) Microbiology 161:719-728.
Another protein, NusA, slows RNAP, in contrast to NusG which increases its processivity, as reflected by the length of the RNA made before RNAP falls off.
Eukaryotic RNA Processing
In multicellular organisms, almost every cell contains the same genome, yet complex spatial and temporal diversity is observed in gene transcripts. This is achieved through multiple levels of processing leading from gene to protein, of which RNA processing is an essential stage. Following the transcription of a gene by RNA polymerases to produce a primary mRNA transcript, further processing is required to produce a stable and functional mature RNA product. This involves various processing steps including RNA cleavage at specific sites, intron removal, called splicing, which substantially increases the transcript repertoire, and the addition of a 5'CAP. Another crucial feature of the RNA processing of most genes is the generation of 3′ ends through an initial endonucleolytic cleavage, followed in most cases by the addition of a poly(A) tail, a process termed 3′ end cleavage and polyadenylation (CPA).
Cleavage and 3'-Polyadenylation (CPA)
Polyadenylation is a required step for the correct termination of nearly all mRNA transcripts. Except for replication-dependent histone genes, metazoan protein-encoding mRNAs contain a uniform 3' end consisting of a stretch of adenosines. In addition to determining the correct transcript length at transcription termination, the poly(A) tail helps to ensure the translocation of the nascent RNA molecule from the nucleus to the cytoplasm, enhances translation efficiency, acts as a signal feature for RNA degradation, and thereby contributes to the production efficiency of a protein.
Cleavage and polyadenylation (CPA) are carried out by the cleavage/polyadenylation apparatus (CPA), a multi-subunit 3′ end processing complex, which involves over 80 proteins, comprised of four core protein subcomplexes, as shown in Figure \(\PageIndex{2}\). These consist of
- cleavage and polyadenylation specificity factor (CPSF), comprised of proteins CPSF1-4, factor interacting with PAPOLA and CPSF1 (FIP1L1), and WD repeat domain 33 (WDR33) (shown in green below);
- cleavage stimulation factor (CstF), a trimer of CSTF1-3 (shown in red below;
- cleavage factor I (CFI), a tetramer of two small nudix hydrolase 21 (NUDT21) subunits, and two large subunits of CPSF7 and/or CPSF6 (shown in orange in Figure 26.2.2 A); Note: Nudix are named for nucleoside diphosphates hydroxylases.
- cleavage factor II (CFII), composed of cleavage factor polyribonucleotide kinase subunit 1 (CLP1) and PCF11 cleavage and polyadenylation factor subunit (PCF11) (shown in yellow in Figure 26.2.2 A). Additional factors include symplekin, the poly(A) polymerase (PAP), and the nuclear poly(A) binding proteins such as poly(A) binding protein nuclear 1 (PABPN1).
CPA is initiated by this complex recognizing specific sequences within the nascent pre-mRNA transcripts termed polyadenylation signals (PAS). The PAS sequence normally consists of either a canonical 6 base sequence, the AATAAA hexamer, or a close variant usually differing by a single nucleotide (e.g., ATTAAA, TATAAA). It is located 10 to 35 nucleotides upstream of the cleavage site (CS) usually consisting of a CA dinucleotide. The PAS is also determined by surrounding auxiliary elements, such as upstream U-rich elements (USE), or downstream U-rich and GU-rich elements and G-rich sequences (DSE).
As soon as the nascent RNA molecule emerges from RNA polymerase II (RNA Pol II), the CPSF complex is recruited to the PAS AATAAA hexamer, through numerous interactions. Upon successful assembly of this macromolecular machinery, CPSF3 performs the endonucleolytic cleavage followed by a non-templated addition of approximately 50-100 A residues.
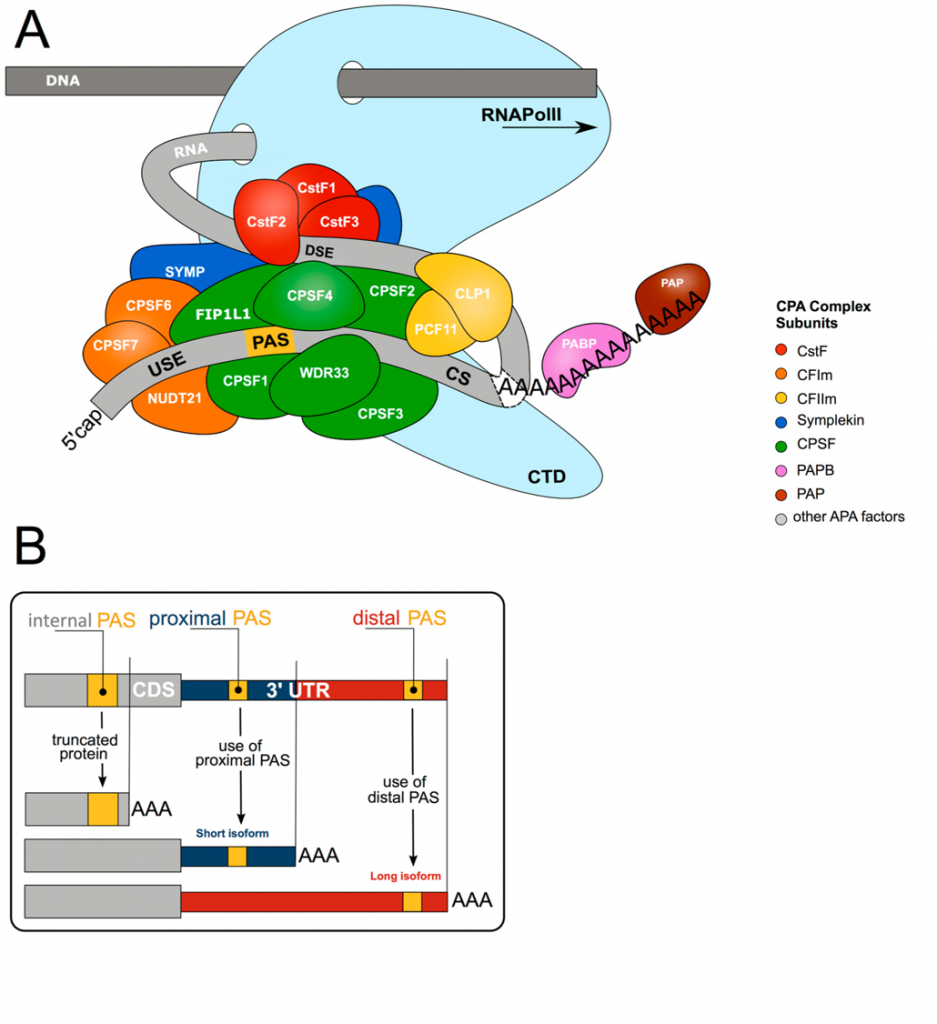
Panel (A) shows the core 3′ end processing machinery consists of complexes composed of multiple trans-acting proteins interacting with RNA via multiple cis-elements (USE = upstream sequence element; PAS = poly(A) signal; CS = cleavage site; DSE = downstream sequence element; CTD = C-terminal domain). Upon co-transcriptional assembly of these complexes, RNA cleavage and polyadenylation occur to form the 3′ end of the nascent RNA molecule.
Panel (B) shows more than 70% of all genes harbor more than one polyadenylation signal (PAS). This gives rise to transcript isoforms differing at the mRNA 3′ end. While alternative polyadenylation (APA) in 3′UTR changes the properties of the mRNA (stability, localization, translation), internal PAS usage (in introns or the coding sequence (CDS)) changes the C-termini of the encoded protein, resulting in different functional or regulatory properties.
Alternative polyadenylation (APA) occurs when more than one PAS is present within a pre-mRNA and provides an additional level of complexity in CPA-mediated RNA processing (Figure 26.2.2 B). Early studies revealed a significant portion of genes undergo APA, and with the advent of next-generation RNA sequencing technologies, the large-scale regulation of genes has become apparent, with approximately 70% of the transcriptome exhibiting APA regulation. As APA determines 3′UTR content and thus the regulatory features available to the mRNA, changes in the APA profile of a gene can have enormous impacts on expression.
For those trying to understand the structure and mechanism of the cleavage/polyadenylation apparatus (CPA), it is especially frustrating that different names are given to the constituents that comprise it, especially when comparing the proteins from different organisms. Hence it is useful to see multiple representations of the complex. Figure \(\PageIndex{3}\) shows a different cartoon representation of the cleavage and polyadenylation reactions. Note again the number of colored subcomplexes within the CPA as well as the different abbreviations shown for the individual proteins. This cartoon diagram is useful in visualizing the different steps involved.

Figure \(\PageIndex{3}\): Cis-regulatory sequence elements and protein factors involved in cleavage and polyadenylation. Marsollier, A.-C.; Joubert, R.; Mariot, V.; Dumonceaux, J. Targeting the Polyadenylation Signal of Pre-mRNA: A New Gene Silencing Approach for Facioscapulohumeral Dystrophy. Int. J. Mol. Sci. 2018, 19, 1347. https://doi.org/10.3390/ijms19051347. Creative Commons Attribution License
Panel (A) shows that the specificity and efficiency of 3′end processing are determined by the binding of more than 80 RNA-binding proteins to regulatory cis-acting RNA sequence elements including the polyadenylation signal (PAS) A[A/U]UAAA; the cleavage site (represented by NN) and the downstream sequence element (DSE). Auxiliary sequences can be found near the polyadenylation signal or the DSE. The core processing complex, which is sufficient for the cleavage and polyadenylation, is composed of approximately 20 proteins, distributed in 8 complexes: the cleavage and polyadenylation specificity factor (CPSF), the cleavage stimulation factor (CstF); the mammalian cleavage factors I (CFIm) and the mammalian cleavage factors II (CFIIm); the single protein poly(A) polymerase (PAP); the single protein poly(A)-binding protein nuclear 1 (PABPN1); the single protein RNA polymerase II large subunit (Pol II); and the symplekin. Subunits of the different factors are indicated.
Panel (B) shows how CPSF and CstF are co-transcriptionally recruited to the poly(A) signal and the DSE respectively, causing an endonucleolytic cleavage of the pre-mRNA between the PAS and the DSE at the cleavage site. Two fragments are generated: one fragment with a free 5′phosphate group which is rapidly degraded by exoribonucleases and one fragment with a free 3′hydroxyl group on which 250 adenines will be added by PAP. The newly-synthetized poly(A) tail is covered by PAPBN1, allowing mRNA circularization and stabilization.
Now let's look at the structure of some of the complexes of the cleavage/polyadenylation apparatus (CPA).
In yeast, the 3' processing is carried out by the cleavage and polyadenylation factor (CPF) which is called the CPSF in humans. On endonuclease cleavage, the RNA bound to RNA polymerase II is in two pieces, as shown in the figure above. The main mRNA now has a 3'-OH which is the site of polyadenylation. The minor cleavage fragment has a 5'-phosphate which gets degraded by the exonuclease Rat1, which as it cleaves the minor product helps displaces RNA polymerase II and helps to stop transcription.
In yeast, the CPF has 14 subunits with polymerase, nuclease, and phosphatase subcomplexes or "modules". The polymerase module, as the name implies, has the poly(A) polymerase, Pap1. Table \(\PageIndex{1}\) below shows some components of the polymerase module of both yeast CPF and human CPSF.
| yeast Polymerase Module of CPF |
Human mammalian polyadenylation specificity factor (mPSF) or CPSF |
| Cft1 | CPSF160 |
| Pfs2 | WDR33 |
| Yth1 - RNA binding subunit | CPSF30 - RNA binding subunit |
| Fip1 - Pap1 binding subunit | FIP1 - Pap1 binding subunit |
Table \(\PageIndex{1}\) Some components of the polymerase module of both yeast CPF and human CPSF.
The nuclease module has an endonuclease (Ysh1) and a Mpe1 protein, which facilitate the cleavage site selection and polyadenylation. Table \(\PageIndex{2}\) below shows some components of the polymerase module in yeast and humans
| yeast | human |
| endonuclease Ysh1 | endonuclease CPSF73 |
| pseudo-nuclease Cft2 | pseudo-nuclease CPSF100 |
| multidomain protein Mpe1 | multidomain protein RBBP6 |
Table \(\PageIndex{2}\): Some components of the polymerase module in yeast and humans
Part of the Cft2 called the yeast polymerase module interacting motif (yPIM), as its name implies, interacts with the polymerase module, in part through the interaction of key and conserved aromatic residues in it (F537, Y549, and F558) with a hydrophobic binding site in Cft1 and Pfs1. These interactions are key in activating and regulating the endonuclease and polyadenylation activities and hence controlling the termination of transcription.
Figure \(\PageIndex{4}\) shows an interactive iCn3D model of the yeast cleavage and polyadenylation specificity factor (CPF) polymerase module in complex with Mpe1, the yPIM of Cft2 and the pre-cleaved CYC1 RNA (7ZGR)
_polymerase_modulewith_Mpe1_yPIM_of_Cft2_pre-cleaved_CYC1_RNA_(7ZGR).png?revision=1&size=bestfit&width=372&height=508)
Figure \(\PageIndex{4}\): Yeast cleavage and polyadenylation specificity factor (CPF) polymerase module in complex with Mpe1, the yPIM of Cft2 and the pre-cleaved CYC1 RNA (7ZGR). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...ePGK2jiiuum7A7
The different parts of the complex are colored as shown below.
- Yth1 (mRNA 3' processing protein): magenta
- CFT1: green
- MPE1: orange
- Polyadenylation subunit 2 (Pfs2): yellow
- cleavage factor 2 protein (CF2P): cyan
- precleaved RNA CYC1: gray spacefill (backbone) with CPK-colored bases
5'-CAP Formation
In eukaryotes, the 5′ cap, found on the 5′ end of the eventual mRNA molecule, consists of a guanine nucleotide connected to the mRNA via an unusual 5′ to 5′ triphosphate linkage, as shown in Figure \(\PageIndex{5}\). This guanosine is methylated on the 7 position directly after capping in vivo by a methyltransferase. It is referred to as the 7-methylguanylate cap, abbreviated m7G.
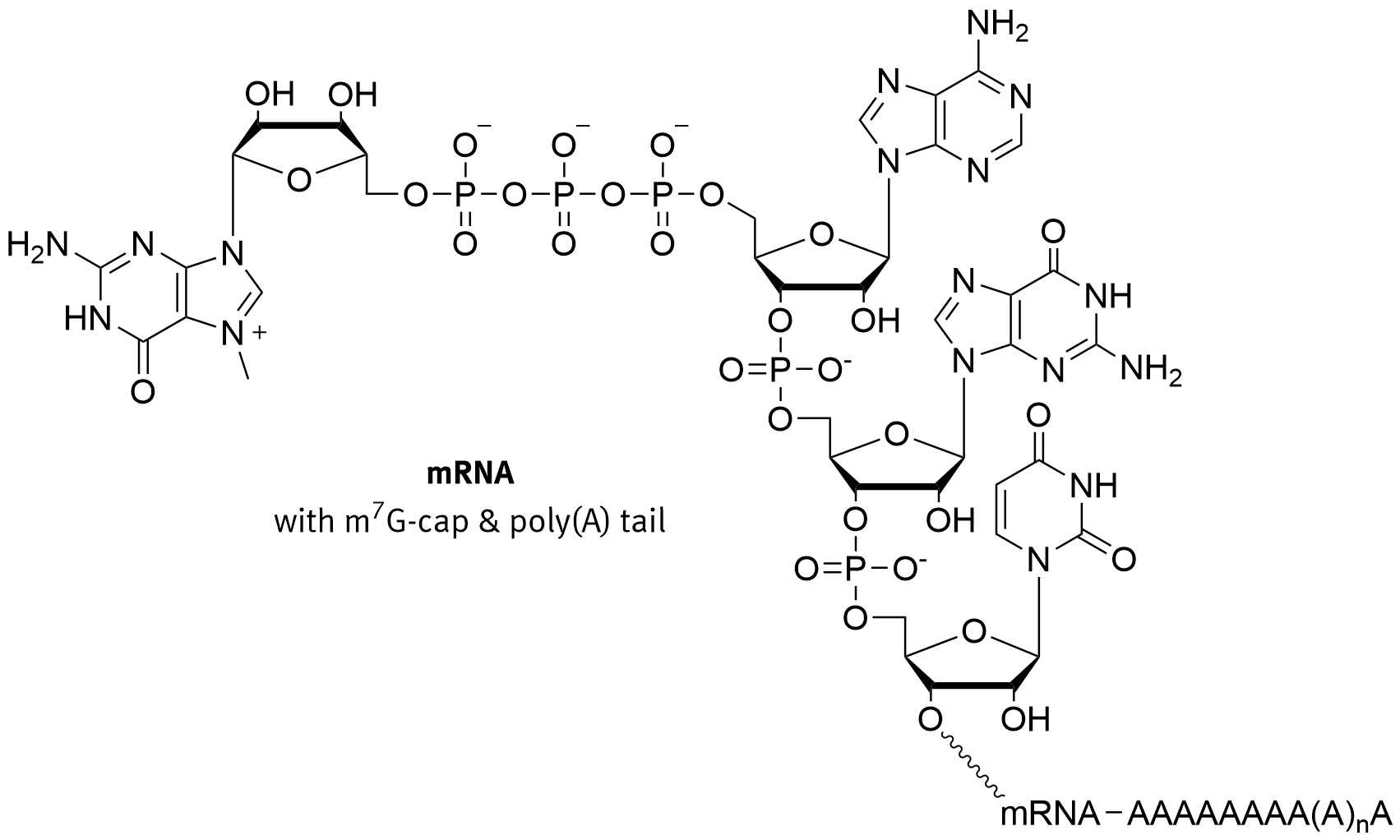
Figure \(\PageIndex{5}\): Structure of the 7-methylguanylate CAP. Brisbane
In multicellular eukaryotes and some viruses, further modifications exist, including the methylation of the 2′ hydroxy-groups of the first 2 ribose sugars of the 5′ end of the mRNA. Cap-1 has a methylated 2′-hydroxy group on the first ribose sugar, while cap-2 has methylated 2′-hydroxy groups on the first two ribose sugars. The 5′ cap is chemically similar to the 3′ end of an RNA molecule (the 5′ carbon of the cap ribose is bonded, and the 3′-OH unbonded). This provides significant resistance to 5′ exonucleases.
Small nuclear RNAs (snRNAs) contain unique 5′-caps. Sm-class snRNAs are found with 5′-trimethylguanosine caps, while Lsm-class snRNAs are found with 5′-monomethylphosphate caps. In bacteria, and potentially also in higher organisms, some RNAs are capped with NAD+, NADH, or 3′-dephospho-coenzyme A. In all organisms, mRNA molecules can be decapped in a process known as messenger RNA decapping.
For capping with 7-methylguanylate, the capping enzyme complex (CEC) binds to RNA polymerase II before transcription starts. As soon as the 5′ end of the new transcript emerges from RNA polymerase II, the CEC carries out the capping process (this kind of mechanism ensures capping, as with polyadenylation). The enzymes for capping can only bind to RNA polymerase II that is engaging in mRNA transcription, ensuring the specificity of the m7G cap almost entirely to mRNA.
The 5′ cap has four main functions:
- Regulation of nuclear export
- Prevention of degradation by exonucleases
- Promotion of translation (see ribosome and translation)
- Promotion of 5′ proximal intron excision
In addition to the polyA tail, the nuclear export of RNA is regulated by the cap-binding complex (CBC), which binds to 7-methylguanylate-capped RNA, as shown in Figure \(\PageIndex{6}\). The CBC is then recognized by the nuclear pore complex and the mRNA is exported. Once in the cytoplasm after the pioneer round of translation, the CBC is replaced by the translation factors eIF4E and eIF4G of the eIF4F complex. This complex is then recognized by other translation initiation machinery including the ribosome, aiding in translation efficiency.

Panel (a) CBC is required for pre-mRNA processing. The co-transcriptional binding of CBC to 7mG prevents the decapping activities of pre-mRNA degradation complexes [DXO (decapping exoribonuclease) and Dcp (decapping mRNA) Xrn2 (5′–3′ exoribonuclease 2)] and promotes pre-mRNA processing. CBC recruits P-TEFb [Cdk9/Cyclin T1 (CycT1)] to transcription initiation sites of specific genes promoting phosphorylation of the RNA pol II CTD at Ser2 residues. This results in the recruitment of splicing factors including SRSF1, which regulates both constitutive and alternative splicing events. Furthermore, CBC interacts with splicing machinery components that result in the spliceosomal assembly. CBC interacts with NELF and promotes pre-mRNA processing of replication-dependent histone transcripts.
Panel (b) CBC forms a complex with Ars2 and promotes miRNA biogenesis by mediating pri-miRNA processing.
Panel (c) CBC/Ars2 promotes pre-mRNA processing of replication-dependent histone transcripts.
Panel (d) CBC promotes the export of U snRNA. CBC interacts with PHAX, which recruits export factors including CRM1 and RAN·GTP.
Panel (e) CBC promotes the export of mRNA. For the export of transcripts over 300 nucleotides, hnRNP C interacts with CBC and inhibits the interaction between CBC and PHAX, allowing the CBC to interact with TREX and the transcript to be translocated to the cytoplasm. CBC interacts with the PARN deadenylase and inhibits its activity, protecting mRNAs from degradation.
Panel (f) CBC mediates the pioneer round of translation. Cbp80 interacts with CTIF, which recruits the 40S ribosomal subunit via eIF3 to the 5′ end of the mRNA for translation initiation. Upon binding of importin-β (Imp-β) to importin-α (Imp-α), mRNA is released from CBC and binds to eIF4E for the initiation of the standard mode of translation. CBC-bound mRNP components not found in eIF4E-bound mRNPs are CTIF, exon junction complex (EJC), and PABPN1.
Panel (g) The standard mode of translation is mediated by eIF4E cap-binding protein. eIF4E is a component of the eIF4F complex which promotes translation initiation.
Capping with 7-methylguanylate prevents 5′ degradation in two ways. First, degradation of the mRNA by 5′ exonucleases is prevented by functionally looking like a 3′ end. Second, the CBC and eIF4E/eIF4G block the access of decapping enzymes to the cap. This increases the half-life of the mRNA, essential in eukaryotes as the export and translation processes take significant time.
The mechanism that promotes the 5′ proximal intron excision during splicing is not well understood, but the 7-methylguanylate cap appears to loop around and interact with the spliceosome, potentially playing a role in the splicing process.
The decapping of a 7-methylguanylate-capped mRNA is catalyzed by the decapping complex made up of at least Dcp1 and Dcp2, which must compete with eIF4E to bind the cap. Thus the 7-methylguanylate cap is a marker of an actively translating mRNA and is used by cells to regulate mRNA half-lives in response to new stimuli. During the decay process, mRNAs may be sent to P-bodies. P-bodies are granular foci within the cytoplasm that contain high levels of exonuclease activity.
Triphosphatase and guanylyltransferase
In capping the new mRNA, three different enzymes act sequentially:
- A phosphatase cleaves a terminal phosphate from the 5' end which has 3 phosphates at the start leaving 2 phosphates (the yeast triphosphatase is called Cet1);
- a GMP is added to the remaining diphosphate on the 5'-end to form a triphosphate in a reverse direction as shown in Figure 5 above (in yeast the RNA guanylyltransferase is called Ceg1);
- a methyl group is added to the N7 of the guanine base by a methylase also called a methyl transferase
These three enzymes are localized to a part of RNA polymerase that is highly phosphorylated, positioning them at the correct location for Cap formation. In yeast, the
Figure \(\PageIndex{7}\) shows an interactive iCn3D model of the Saccharomyces cerevisiae Cet1 (the triphosphatase)-Ceg1 (the guanylyltransferase) mRNA Capping Apparatus (3KYH)
.png?revision=1&size=bestfit&width=594&height=371)
Figure \(\PageIndex{7}\): Saccharomyces cerevisiae Cet1-Ceg1 mRNA Capping Apparatus (3KYH) . (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...UWQ32mQrcF1eN6
The two enzymes exist as a heterotetramer of two homodimers. Two beta (Cet1-triphosphatase) subunits are shown in gray and the two alpha (Ceg1-guanylyltransferase) subunits are shown in cyan. The active site residues are shown in CPK-colored sticks and labeled. The two enzymes interact with yeast RNAP II mostly through Ceg1. Specifically, the Ceg1 oligonucleotide domain interacts with a motif, WAQKW (247-251), on Cet1. A conformational change in a flexible linker after that motif allows capping to ensue.
Methyl transferase
The next step is the methyl transfer to the N7 of guanine in the 5' end of the cap. Figure \(\PageIndex{8}\) shows an interactive iCn3D model of the Structure of a bacterial mRNA cap (Guanine-n7) methyltransferase (1RI1)
_methyltransferase_(1RI1)_with_bound_S-adenoysl-homocysteine.png?revision=1&size=bestfit&width=494&height=403)
Figure \(\PageIndex{8}\): Structure of a bacterial mRNA cap (Guanine-n7) methyltransferase (1RI1) with bound S-adenoysl-homocysteine. (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...YfaJ6W93zSDaX7
7-Methyl-guanosine-5'-triphosphate-5'-guanosine (GTG) is shown in sticks. The N7 nitrogen of guanine is labeled. S-Adenoslyl-L-homocysteine, the leaving group after methylation by S-adenosyl-L-methionine (SAM), is shown in spacefill. Key amino acid side chains in the active site are labeled (in small letters). The structure is most consistent with an in-line methyl transfer from SAM to the attacking nucleophile, the N7 of guanine. Specificity in most capping methylases occurs through noncovalent interactions (mostly base stacking, with two amino acid aromatic groups (an example of π-π stacking), or one aromatic and one nonpolar side chain. In the bacterial example shown above, the interactions include hydrophobic and hydrogen bonding interactions using Y284, F24, P175, E225, H144, and Y145.
Decapping
"What goes up must come down!" If the mRNA is capped during synthesis, there must be an enzyme to decap it. Figure \(\PageIndex{9}\) shows an interactive iCn3D model of the yeast mRNA decapping enzyme Dcp1-Dcp2 complex in the ATP bound closed conformation (2QKM)
.png?revision=1&size=bestfit&width=491&height=372)
Figure \(\PageIndex{9}\): Yeast mRNA decapping enzyme Dcp1-Dcp2 complex in the ATP bound closed conformation (2QKM). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...hBPokeNeMfcGUA
The rendering is colored as shown below:
- a chain in cyan
- b chain in gray
- nudix motif in magenta
- ATP in spacefill
Dcp2p, as with many other enzymes, has an open (inactive) and closed (active) conformation suggesting that a conformational change between the two states regulates decapping. The ATP binding site demarcates the active site. the Dcp1 protein probably functions to stabilize the closed state.
mRNA Splicing
Eukaryotic genes that encode polypeptides are composed of coding sequences called exons (ex-on signifies that they are expressed) and intervening sequences called introns (int-ron denotes their intervening role). Transcribed RNA sequences corresponding to introns do not encode regions of the functional polypeptide and are removed from the pre-mRNA during processing. All of the intron-encoded RNA sequences must be completely and precisely removed from a pre-mRNA before protein synthesis so that the exon-encoded RNA sequences are properly joined together to code for a functional polypeptide. If the process errs by even a single nucleotide, the sequences of the rejoined exons would be shifted, and the resulting polypeptide would be nonfunctional. The process of removing intron-encoded RNA sequences and reconnecting those encoded by exons is called RNA splicing. Intron-encoded RNA sequences are removed from the pre-RNA while it is still in the nucleus. Although they are not translated, introns appear to have various functions, including gene regulation and mRNA transport. On completion of these modifications, the mature transcript, the mRNA that encodes a polypeptide, is transported out of the nucleus, destined for the cytoplasm for translation. Introns can be spliced out differently, resulting in various exons being included or excluded from the final mRNA product. This process is known as alternative splicing. The advantage of alternative splicing is that different types of mRNA transcripts can be generated, all derived from the same DNA sequence. In recent years, it has been shown that some archaea also can splice their pre-mRNA.
The splicing reaction is catalyzed by the spliceosome, a macromolecular complex formed by five small nuclear ribonucleoproteins (snRNPs), termed U1, U2, U4, U5, and U6, and approximately 200 proteins, as shown in Figure \(\PageIndex{10}\). Each of these snRNPs contains snRNAs that can interact with each other through intrastrand hydrogen bonding and hence which help localize the snRNPs to the complexes. The assembly of the spliceosome on pre-mRNA includes the binding of U1 snRNP, U2 snRNP, the pre-formed U4/U6-U5 triple snRNP, and the Prp19 complex. This assembly occurs through the recognition of several sequence elements on the pre-mRNA that define the exon/intron boundaries, which include the 5′ and 3′ splice sites (SS), the associated 3′ sequences for intron excision, the polypyrimidine (Py) tract, and the branch point sequence (BPS). The assembly of the spliceosome during the process is depicted in Figure \(\PageIndex{10}\).

In the first step of the splicing process, the 5′ splice site (GU, 5′ SS) is bound by the U1 snRNP, and the splicing factors SF1/BBP and U2AF cooperatively recognize the branch point sequence (BPS), the polypyrimidine (Py) tract, and the 3′ splice site (AG, 3′ SS) to assemble complex E. The binding of the U2 snRNP to the BPS results in the pre-spliceosomal complex A. Subsequent steps lead to the binding of the U4/U5–U6 tri-snRNP and the formation of complex B. Complex C is assembled after rearrangements that detach the U1 and U4 snRNPs to generate complex B*. Complex C is responsible for the two transesterification reactions at the SS. Additional rearrangements result in the excision of the intron, which is removed as a lariat RNA, and ligation of the exons. The U2, U5, and U6 snRNPs are then released from the complex and recycled for subsequent rounds of splicing
There are different ways that pre-spliceosomes convert to full spliceosomes. The interactions of the small ribonucleoproteins U1 and U2, which bind at the 5' and 3' end of the introns respectively, are key. When multiple introns exist, a few different pathways occur. The interactions can be upstream (cross-introns) and downstream (cross-exons), as shown in Figure \(\PageIndex{11}\).

Figure \(\PageIndex{11}\): Implications of synergistic U2 recruitment for the mechanism of exon and intron recognition. Braun et al. (2018) . Synergistic assembly of human pre-spliceosomes across introns and exons. eLife 7:e37751. https://doi.org/10.7554/eLife.37751. Creative Commons Attribution License
The left-hand side of the figure shows how the interaction of U1 and U2 can occur across a single intron, to form the mature version of the spliceosome showing the U1-U2 interaction. The colored arrow shows possible interactions that either speed up or slow down the U1-U2 mature interactions.
The right-hand side shows the formation of an intermediate cross-exon U2-U1 pair in which the exons that flank two introns are first delineated, which then leads to the physical U1-U2 interaction in the mature spliceosome when cross-intron interactions occur.
Now let's move to show the actual structures of two of the spliceosome structures shown in Figure 10 above. Figure \(\PageIndex{12}\) shows an interactive iCn3D model of the human fully-assembled precatalytic spliceosome (pre-B complex) (6QX9)
_(6QX9).png?revision=1&size=bestfit&width=607&height=533)
Figure \(\PageIndex{12}\): Human fully-assembled precatalytic spliceosome (pre-B complex) (6QX9). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zLLvMh82m5u9M8
The full structure of the spliceosome is almost too complicated to understand even if displayed in a molecular model. It's better grasped in a way by the cartoon figure shown previously. To avoid unnecessary visual complexity, the protein subunits in the iCn3D model of the human spliceosome are shown in a gray cartoon, and only the RNA molecules in the Un small ribonucleoproteins are shown in color, as described below. The resolution is such that each nucleotide in the RNA polymers is shown just as a single-colored sphere not connected to the next nucleotide in the RNA.
- U1 snRNA - magenta
- U6 snRNA - orange
- U5 snRNA - yellow
- U2 snRNA - cyan
- U4 snRNA - blue
- AdML pre-mRNA - black
Since it has U1, U2, U4, U5, and U6, it most closely resembles Complex B (or an immediate precursor pre-B of it) as shown in Figure 10. This form occurs before U1 snRNP dissociates. A helix from the 5'-single-stranded U1 snRNA inserts into a helicase in the complex between two RecA domains which bind ATP and unfold the nucleic acids. This allows the 5' single-stranded RNA to form interactions with an ACAGAGA sequence (mobile loop) in U6. These conformational changes allow the eventual separation of the U4 and U6 snRNA, freeing the snRNA in U6 is form the catalytic site. The B complex itself does not have a functioning active site. After the dissociation of U1 and U4 snRNPs, and the binding of another 20 or so proteins, the active spliceosome is formed.
Now let's look at the final catalytic structure, Complex C, which contains only three of the snRibonucleoproteins, U2, U5, and U6. Figure \(\PageIndex{13}\) shows an interactive iCn3D model of the Human C Complex Spliceosome (7A5P)
.png?revision=1&size=bestfit&width=440&height=465)
Figure \(\PageIndex{13}\): Human C Complex Spliceosome (7A5P). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...5co1nofAVEnyR6
The color coding is as follows:
- protein - gray
- U6 snRNA - orange
- U5 snRNA - yellow
- U2 snRNA - cyan
- AdML pre-mRNA - magenta
The interactions of the snRNAs are key for structure and catalysis in the spliceosome, and in the figures above the interactions are had to discern. Figure \(\PageIndex{14}\) shows the interactions among U2, U5, and U6 snRNA in the human and yeast spliceosomes.

Figure \(\PageIndex{14}\): The RNA elements and the splicing active site of the human mature Bact complex. Zhang, X., Yan, C., Zhan, X. et al. Structure of the human-activated spliceosome in three conformational states. Cell Res 28, 307–322 (2018). https://doi.org/10.1038/cr.2018.14. Creative Commons Attribution 4.0 Unported License. http://creativecommons.org/licenses/by/4.0/
Panel (A) shows the structure of the RNA elements in the core of the human mature Bact complex. The color-coded RNA elements of the human Bact complex are shown in the left panel, and their superposition with those of the S. cerevisiae Bact complex is displayed in the right panel. All yeast RNA elements are colored grey. The helix II of the U2/U6 duplex in the human Bact complex is bent relative to that in the yeast complex.
Panel (B) shows a structural overlay of the active site RNA elements between the human and S. cerevisiae Bact complexes.
Panel (C) shows that the U6/intron duplex in the human Bact complex is considerably longer than that in the S. cerevisiae Bact complex.
In mammals, the first catalytic step of the splicing reaction begins when the U1 snRNP binds the 5′ SS of the intron (defined by the consensus sequence AGGURAGU), and the splicing factors SF1 and U2AF cooperatively recognize the BPS, Py, and 3′ SS to assembled complex E or the commitment complex. Subsequently, U2 snRNP and additional proteins are recruited to the pre-mRNA BPS to form the pre-spliceosome or complex A. The binding of the U4/U6-U5 tri-snRNP forms the pre-catalytic spliceosome or complex B. After RNA-RNA and RNA-protein rearrangements at the heart of the spliceosome, U1 and U4 are released to form the activated complex B or complex B* This complex is responsible for executing the first catalytic step, through which the phosphodiester bond at the 5′ SS of the intron is modified by the 2′-hydroxyl of adenosine of the BPS to form a free 5′ exon and a branched intron, as shown in Figure \(\PageIndex{15}\). The reaction of the 2'-hydroxyl from the branch point adenosine nucleotide is known as a transesterification reaction. During this process, additional rearrangements occur to generate the catalytic spliceosome or complex C (see Figure 10 above), which is responsible for catalyzing the second transesterification reaction leading to intron excision and exon–exon ligation. The resulting intron structure is referred to as a lariat structure. After the second catalytic step, the U2, U5, and U6 snRNPs are released from the post-spliceosomal complex and recycled for additional rounds of splicing.

Panel (A) shows a schematic diagram of the pre-mRNA with exons and introns indicated. Key sequences are required for splicing at the 5' and 3' intron locations, and for the recognition and positioning of the branch point Adenosine residue for the first transesterification reaction. (
Panel (B) shows a schematic of the two transesterification reactions required for intron removal. The branch point 2'-OH residue mediates attack on the 5'-phosphate of the intron guanosine residue located at the 5'-splice site. This releases the 3' hydroxyl of Exon 1 which subsequently mediates the attack of the 5' phosphate of the first guanosine residue in Exon 2. The 3' hydroxyl of the intron guanine residue is released forming the Lariat structure and Exon 1 is ligated to Exon 2.
Alternative Splicing (AS) offers an additional mechanism for regulating protein production and function. AS options are determined by the expression of or exposure to trans elements present within unique cellular locations and environments. Additional sequence elements within the mRNA, known as exonic and intronic splicing silencers or enhancers (ESS, ISS, ESE, and ISE, respectively), participate in the regulation of AS. Specific RNA-binding proteins, including heterogeneous nuclear ribonucleoproteins (hnRNPs) and serine/arginine-rich (SR) proteins, recognize these sequences to positively or negatively regulate AS, as shown in Figure \(\PageIndex{16}\). These regulators, together with an ever-increasing number of additional auxiliary factors, provide the basis for the specificity of this pre-mRNA processing event in different cellular locations within the body.

The core cis sequence elements that define the exon/intron boundaries (5′ and 3′ splice sites (SS), GU-AG, polypyrimidine (Py) tract, and branch point sequence (BPS)) are poorly conserved. Additional enhancer and silencer elements in exons and introns (ESE: exonic splicing enhancers; ESI: exonic splicing silencers; ISE: intronic splicing enhancers; ISI: intronic splicing silencers) contribute to the specificity of AS regulation. Trans-acting splicing factors, such as SR family proteins and heterogeneous nuclear ribonucleoprotein particles (hnRNPs), bind to enhancers and silencers and interact with spliceosomal components. In general, SR proteins bound to enhancers facilitate exon definition, and hnRNPs inhibit this process. These trans-acting elements are expressed differentially within different locations or under different environmental stimuli to regulate AS.
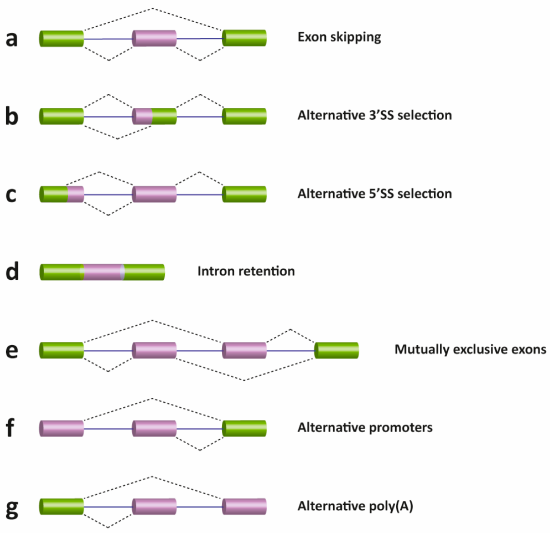
There are several different types of AS events, which can be classified into four main subgroups. The first type is exon skipping, which is the major AS event in higher eukaryotes. In this type of event, a cassette exon is removed from the pre-mRNA as shown in Figure \(\PageIndex{17}\) (panel A). The second and third types are alternative 3′ and 5′ SS selections (panels b and c). These types of AS events occur when the spliceosome recognizes two or more splice sites at one end of an exon. The fourth type is intron retention (panel d), in which an intron remains in the mature mRNA transcript. This AS event is much more common in plants, fungi, and protozoa than in vertebrates. Other events that affect the transcript isoform outcome include mutually exclusive exons (panel e), alternative promoter usage (panel f), and alternative polyadenylation (panel g).
Here is a detailed and incredible narrated animation of splicing and the spliceosome.
References
- Parker, N., Schneegurt, M., Thi Tu, A-H., Lister, P., Forster, B.M. (2019) Microbiology. Openstax. Available at: https://opentextbc.ca/microbiologyopenstax/
- Palazzo, A., and Lee, E.S. (2015) Non-coding RNA: what is function and what is junk? Frontiers in Genetics 6:2 Available at: file:///C:/Users/flatt/AppData/Local/Temp/fgene-06-00002.pdf
- Wikipedia contributors. (2020, July 9). RNA. In Wikipedia, The Free Encyclopedia. Retrieved 15:30, August 6, 2020, from https://en.Wikipedia.org/w/index.php?title=RNA&oldid=966784317
- Burenina, O.Y., Oretskaya, T.S., and Kubareva, E.A. (2017) Non-Coding RNAs As Transcriptional Regulators in Eukaryotes. Acta Naturae 9(4):13-25. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5762824/
- Khatter, H., Vorlander, M.K., and Muller C.W. (2017) RNA polymerase I and III: similar yet unique. Current Opinion in Structural Biology 47:88-94. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X17300313
- Wikipedia contributors. (2020, May 8). Sigma factor. In Wikipedia, The Free Encyclopedia. Retrieved 17:50, August 7, 2020, from https://en.Wikipedia.org/w/index.php?title=Sigma_factor&oldid=955570499
- Bae, B., Felkistov, A., Lass-Napiokowska, A., Landick, R., and Darst, S.A. (2015) Structure of a bacterial RNA polymerase holoenzyme open protomer complex. eLife 4:e08504. Available at: https://elifesciences.org/articles/08504
- Petrenko, N., Jin, Y., Dong, L., Wong, K.H., and Struhl, K. (2019) Requirements for RNA polymerase II preinitiation complex formation in vivo. eLife 8:e43654. Available at: https://elifesciences.org/articles/43654
- Gupta, K., Sari-Ak, D., Haffke, M., Trowitzsch, S., and Berger, I. (2016) Zooming in on transcription preinitiation. J Mol Biol. 428(12):2581-2591. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4906157/
- Wikipedia contributors. (2020, April 17). TATA-binding protein. In Wikipedia, The Free Encyclopedia. Retrieved 14:54, August 8, 2020, from https://en.Wikipedia.org/w/index.php?title=TATA-binding_protein&oldid=951583592
- Patel, A.B., Greber, B.J., and Nogales, E. (2020) Recent insights into the structure of TFIID, its assembly, and its binding to core promoter. Curr Op Struct Bio 61:17-24. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X19301113#fig0010
- Ruff, E.F., Record, Jr., M.T., Artsimovitch, I., (2015) Initial events in bacterial transcription initiation. Biomolecules 5(2):1035-1062. Available at: https://www.mdpi.com/2218-273X/5/2/1035/htm
- Kireeva, M., Opron, K., Seibold, S., Domecq, C., Cukier, R.I., Coulombe, B., Kashlev, M., and Burton, Z. (2102) Molecular dynamics and mutational analysis of the catalytic and translocation cycle of RNA polymerase. BMC Biophysics 5(1):11. Available at: https://www.researchgate.net/publication/225281979_Molecular_dynamics_and_mutational_analysis_of_the_catalytic_and_translocation_cycle_of_RNA_polymerase/figures?lo=1
- Washburn, R.S., and Gottesman, M.E. (2015) Regulation of transcription elongation and termination. Biomolecules 5(2):1063-1078. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4496710/pdf/biomolecules-05-01063.pdf
- Zenkin, N., and Yuzenkova, Y. (2015) New insights into the functions of transcription factors that bind the RNA polymerase secondary channel. Biomolecules 5(3):1195-1209. Available at: https://www.mdpi.com/2218-273X/5/3/1195/htm
- Gocheva, V., LeGall, A., Boudvillain, M., Margeat, E., and Nollmann, M. (2015) Direct observation of the translocation mechanism of transcription termination factor Rho. Nuc Acids Res 43(1):10.1093. Available at: https://www.researchgate.net/publication/272162172_Direct_observation_of_the_translocation_mechanism_of_transcription_termination_factor_Rho
- Miki, T.S., Carl, S.H., and Groβhans, H. (2017) Two distinct transcription termination modes dictated by promoters. Genes & Dev 31:1-10. Available at: https://www.researchgate.net/publication/320350041_Two_distinct_transcription_termination_modes_dictated_by_promoters
- Gurumurthy, A., Shen, Y., Gunn, E.M., Bungert, J. (2018) Phase separation and transcription regulation: Are Super-Enhancers and Locus Control Regions primary sites of transcription complex assembly? BioEssays 1800164. Available at: https://www.researchgate.net/publication/329331157_Phase_Separation_and_Transcription_Regulation_Are_Super-Enhancers_and_Locus_Control_Regions_Primary_Sites_of_Transcription_Complex_Assembly
- Suñé-Pou, M., Prieto-Sánchez, Boyero-Corral, S., Moreno-Castro, C., El Yousfi, Y., Suñé-Negre, J.M., Hernández-Munain, C., and Suñé, C. (2017) Targeting splicing in the treatment of human disease. Genes 8(3):87. Available at: https://www.mdpi.com/2073-4425/8/3/87/htm
- Schaughency, P., Merran, J., and Corden J.L. (2014) Genome-wide mapping of yeast RNA polymerase II termination. PLOS Genetics 10(10):e1004632 Available at: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004632
- Nourse, J., Spada, S., and Danckwardt, S. (2020) Emerging roles of RNA 3'-end cleavage and polyadenylation in pathogenesis, diagnosis, and therapy of human disorders. Biomolecules 10(6):915. Available at: https://www.mdpi.com/2218-273X/10/6/915/htm
- Wikipedia contributors. (2020, July 30). Five-prime cap. In Wikipedia, The Free Encyclopedia. Retrieved 05:53, August 11, 2020, from https://en.Wikipedia.org/w/index.php?title=Five-prime_cap&oldid=970240533
- Cortes, T. and Cox, R.A. (2015) Transcription and translation of the rpsJ, rplN and rRNA operons of the tubercle bacillus. Microbiology (2015) 161:719-728. Available at: https://www.microbiologyresearch.org/docserver/fulltext/micro/161/4/719_mic000037.pdf?expires=1597159574&id=id&accname=guest&checksum=6FFC9C066EF41C7799FAE843CE94C49F
- Hein, P.P. and Landick, R. (2010) The bridge helix coordinates movements of modules in RNA polymerase. BMC Biology 8:141. Available at: https://bmcbiol.biomedcentral.com/articles/10.1186/1741-7007-8-141
- Gonatopoulos-Pournatzis, T., and Cowling, V.H. (2014) Cap-binding complex (CBC). Biochem. J. 457:231-242. Available at: https://www.researchgate.net/publication/259392894_Cap-binding_complex_CBC