
Case Study: Cystic Fibrosis - CER
- Page ID
- 26446
This page is a draft and is under active development.
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Part I: A Case of Cystic Fibrosis
Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency room because she had been suffering from a chronic cough. In addition, they said that Zoey sometimes would "wheeze" a lot more than they thought was normal for a child with a cold. Upon arriving at the emergency room, the attending pediatrician noted that salt crystals were present on Zoey's skin and called Dr. Weyland, a pediatric pulmonologist. Dr. Weyland suspects that baby Zoey may be suffering from cystic fibrosis.
CF affects more than 30,000 kids and young adults in the United States. It disrupts the normal function of epithelial cells — cells that make up the sweat glands in the skin and that also line passageways inside the lungs, pancreas, and digestive and reproductive systems.
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system.
When the CFTR protein is defective, epithelial cells can't regulate the way that chloride ions pass across cell membranes. This disrupts the balance of salt and water needed to maintain a normal thin coating of mucus inside the lungs and other passageways. The mucus becomes thick, sticky, and hard to move, and can result in infections from bacterial colonization.
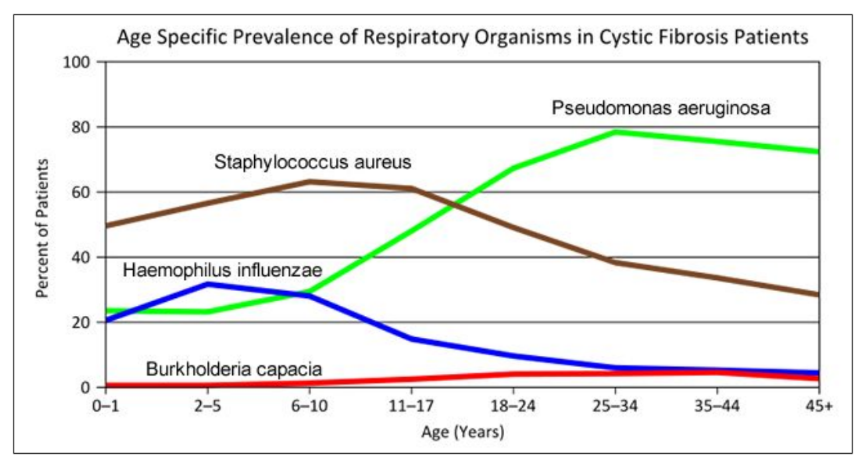
- "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die" This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age have a better chance of survival than babies in the 18th century?
- What symptoms lead Dr. Weyland to his initial diagnosis?
- Consider the graph of infections, which organism stays relatively constant in numbers over a lifetime. What organism is most likely affecting baby Zoey?
- What do you think is the most dangerous time period for a patient with CF? Justify your answer.
Part II: CF is a disorder of the cell membrane.
Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride channel – a cellular doorway for salt ions that is defective in people with cystic fibrosis.
His findings, reported in a series of three recent papers in the Journal of General Physiology, detail the type and order of molecular events required to open and close the gates of the cystic fibrosis chloride channel, or as scientists call it, the cystic fibrosis transmembrane conductance regulator (CFTR).
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel.
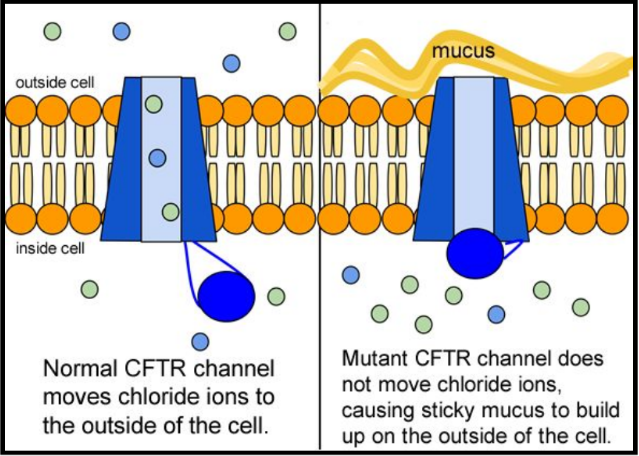
5. Suggest a molecular fix for a mutated CFTR channel. How would you correct it if you had the ability to tinker with it on a molecular level?
6. Why would treatment that targets the CFTR channel not be effective for 2⁄3 of those with cystic fibrosis?
7. Sweat glands cool the body by releasing perspiration (sweat) from the lower layers of the skin onto the surface. Sodium and chloride (salt) help carry water to the skin's surface and are then reabsorbed into the body. Why does a person with cystic fibrosis have salty tasting skin?
Part III: No cell is an island
Like people, cells need to communicate and interact with their environment to survive. One way they go about this is through pores in their outer membranes, called ion channels, which provide charged ions, such as chloride or potassium, with their own personalized cellular doorways. But, ion channels are not like open doors; instead, they are more like gateways with high-security locks that are opened and closed to carefully control the passage of their respective ions.
In the case of CFTR, chloride ions travel in and out of the cell through the channel’s guarded pore as a means to control the flow of water in and out of cells. In cystic fibrosis patients, this delicate salt/water balance is disturbed, most prominently in the lungs, resulting in thick coats of mucus that eventually spur life-threatening infections. Shown below are several mutations linked to CFTR:
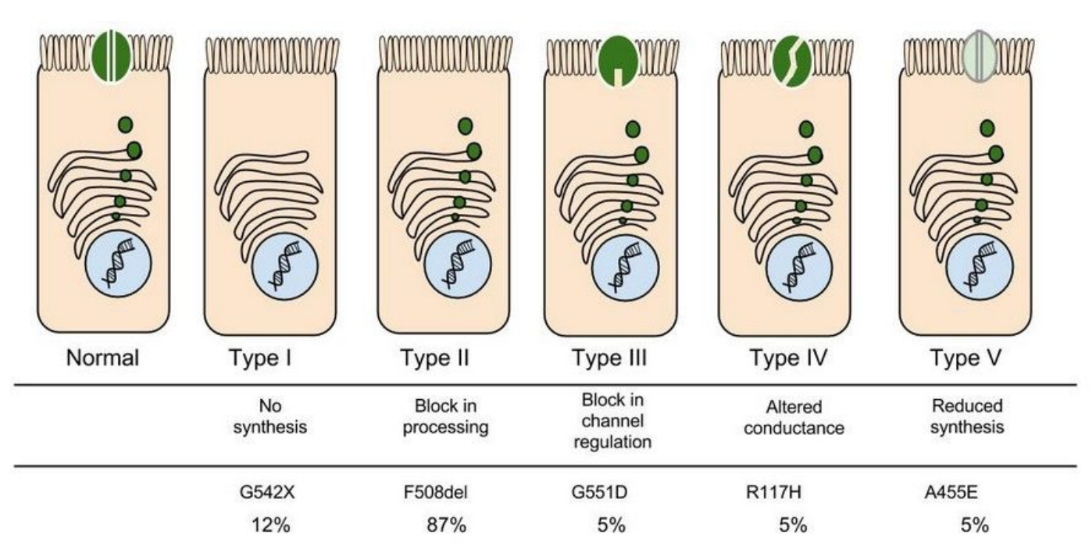
| Mutation | Description |
| Class I | Gene contains a stop signal that prevents CFTR from being made. |
| Class II | CFTR is made, but does not reach the cell membrane. |
| Class III | CFTR is made and in the right place, but does not function normally. |
| Class IV | Channel does not move substances efficiently or at all. |
| Class V | CFTR is made in smaller than normal quantities. |
8. Which mutation do you think would be easiest to correct. Justify your answer.
9. Consider what you know about proteins, why does the “folding” of the protein matter?
Part IV: Open sesame
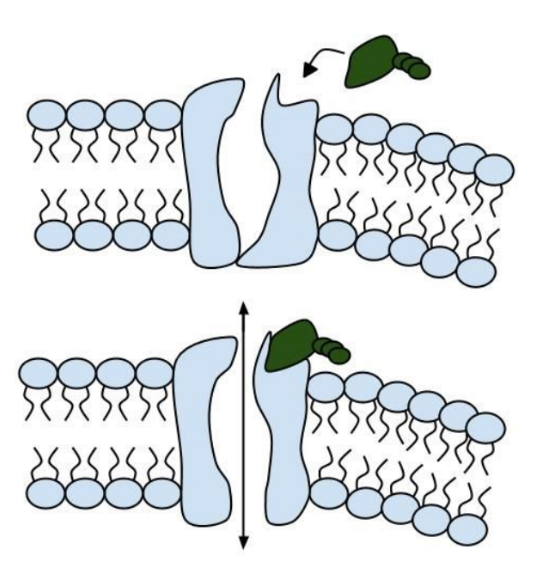
Among the numerous ion channels in cell membranes, there are two principal types: voltage-gated and ligand-gated. Voltage-gated channels are triggered to open and shut their doors by changes in the electric potential difference across the membrane. Ligand-gated channels, in contrast, require a special “key” to unlock their doors, which usually comes in the form of a small molecule.
CFTR is a ligand-gated channel, but it’s an unusual one. Its “key” is ATP, a small molecule that plays a critical role in the storage and release of energy within cells in the body. In addition to binding the ATP, the CFTR channel must snip a phosphate group – one of three “P’s” – off the ATP molecule to function. But when, where and how often this crucial event takes place has remains obscure.
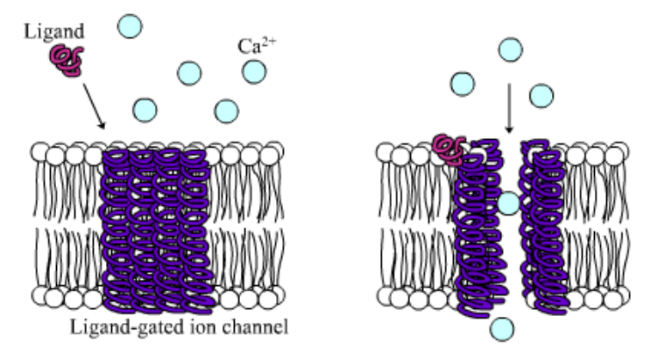
10. Compare the action of the ligand-gated channel to how an enzyme works.
11. Consider the model of the membrane channel, What could go wrong to prevent the channel from opening?
12. Where is ATP generated in the cell? How might ATP production affect the symptoms of cystic fibrosis?
13. Label the image below to show how the ligand-gated channel for CFTR works. Include a summary.
Part V: Can a Drug Treat Zoey’s Condition?
Dr. Weyland confirmed that Zoey does have cystic fibrosis and called the parents in to talk about potential treatments. “Good news, there are two experimental drugs that have shown promise in CF patients. These drugs can help Zoey clear the mucus from his lungs. Unfortunately, the drugs do not work in all cases.” The doctor gave the parents literature about the drugs and asked them to consider signing Zoey up for trials.
The Experimental Drugs
Ivacaftor TM is a potentiator that increases CFTR channel opening time. We know from the cell culture studies that this increases chloride transport by as much as 50% from baseline and restores it closer to what we would expect to observe in wild type CFTR. Basically, the drug increases CFTR activity by unlocking the gate that allows for the normal flow of salt and fluids.
In early trials, 144 patients all of whom were age over the age of 12 were treated with 150 mg of Ivacaftor twice daily. The total length of treatment was 48 weeks. Graph A shows changes in FEV (forced expiratory volume) with individuals using the drug versus a placebo. Graph B shows concentrations of chloride in patient’s sweat.
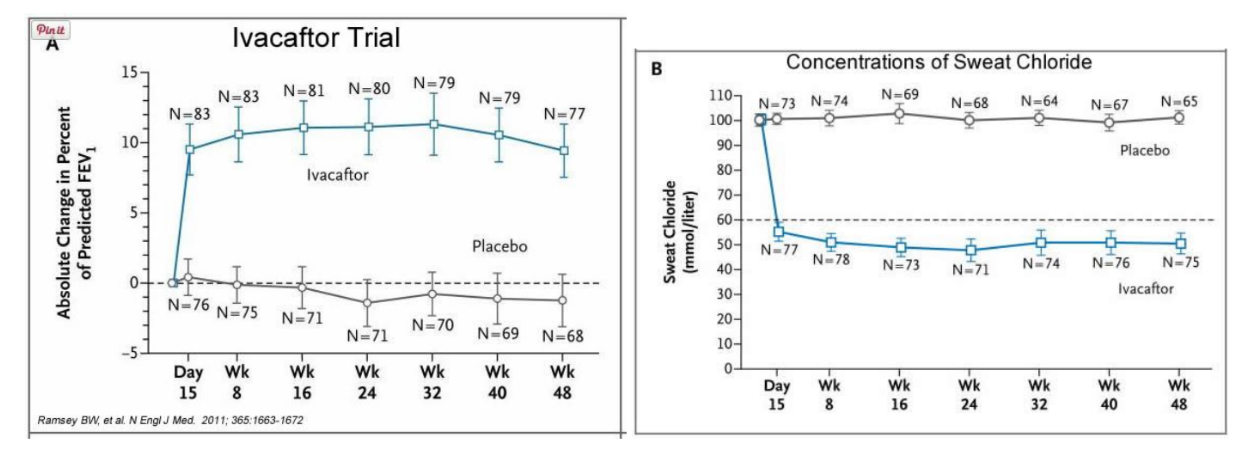
14. What is FEV? Describe a way that a doctor could take a measurement of FEV.
15. Why do you think it was important to have placebos in both of these studies?
16. Which graph do you think provides the most compelling evidence for the effectiveness of Ivacafor?
Defend your choice.
17. Take a look at the mutations that can occur in the cell membrane proteins from Part III. For which mutation do you
think Ivacaftor will be most effective? Justify your answer.
18. Would you sign Zoey up for clinical trials based on the evidence? What concerns would a parent have before
considering an experimental drug?
Part VI: Zoey’s Mutation
Dr. Weyland calls a week later to inform the parents that genetic tests show that Zoey chromosomes show that she has two copies of the F508del mutation. This mutation, while the most common type of CF mutation, is also one that is difficult to treat with just Ivacaftor. There are still some options for treatment.
Lumacaftor
In people with the most common CF mutation, F508del, a series of problems prevents the CFTR protein from taking its correct shape and reaching its proper place on the cell surface. The cell recognizes the protein as not normal and targets it for degradation before it makes it to the cell surface. In order to treat this problem, we need to do two things: first, an agent to get the protein to the surface, and then ivacaftor (VX-770) to open up the channel and increase chloride transport. VX-809 has been identified as a way to help with the trafficking of the protein to the cell surface. When added VX-809 is added to ivacaftor (now called Lumacaftor,) the protein gets to the surface and also increases in chloride transport by increasing channel opening time.
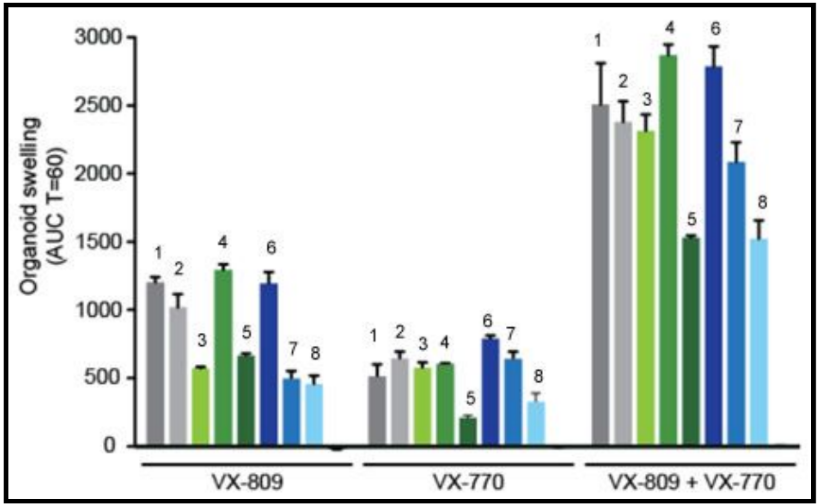
In early trials, experiments were done in-vitro, where studies were done on cell cultures to see if the drugs would affect the proteins made by the cell. General observations can be made from the cells, but drugs may not work on an individual’s phenotype. A new type of research uses ex-vivo experiments, where rectal organoids (mini-guts) were grown from rectal biopsies of the patient that would be treated with the drug. Ex-vivo experiments are personalized medicine, each person may have different correctors and potentiators evaluated using their own rectal organoids. The graph below shows how each drug works for 8 different patients (#1-#8)
19. Compare ex-vivo trials to in-vitro trials.
20. One the graph, label the group that represents Ivacaftor and Lumacaftor. What is the difference between these two drugs?
21. Complete a CER Chart. If the profile labeled #7 is Zoey, rank the possible drug treatments in order of their effectiveness for her mutation. This is your CLAIM. Provide EVIDENCE to support your claim. Provide REASONING that explains why this treatment would be more effective than other treatments and why what works for Zoey may not work for other patients. This is where you tie the graph above to everything you have learned in this case. Attach a page.