
11.10: Genomic Libraries
- Page ID
- 42825

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A genomic library might be a tube full of recombinant bacteriophage. Each phage DNA molecule contains a fragmentary insert of cellular DNA from a foreign organism. The library is made to contain a representation of all of possible fragments of that genome. Bacteriophage are often used to clone genomic DNA fragments because:
- phage genomes are bigger than plasmids and can be engineered to remove a large amount of DNA that is not needed for infection and replication in bacterial host cells.
- the missing DNA can thus be replaced by foreign insert DNA fragments as long as 18- 20kbp (kilobase pairs), nearly 20X as long as typical cDNA inserts in plasmids.
- purified phage coat proteins can be mixed with the recombined phage DNA to make infectious phage particles that would infect host bacteria, replicate lots of new recombinant phage, and then lyse the cells to release the phage.
The need for vectors like bacteriophage that can accommodate long inserts becomes obvious from the following bit of math. A typical mammalian genome consists of more than 2 billion base pairs. Inserts in plasmids are very short, rarely exceeding 1000 base pairs. Dividing 2,000,000,000 by 1000, you get 2 million, a minimum number of phage clones that must be screened to find a sequence of interest. In fact, you would need many more than this number of clones to find a gene (or parts of one!). Of course, part of the solution to this “needle in a haystack” dilemma is to clone larger DNA inserts in more accommodating vectors.
From this brief description, you may recognize the common strategy for genetically engineering a cloning vector: determine the minimum properties that your vector must have and remove non-essential DNA sequences. Consider the Yeast Artificial Chromosome (YAC), hosted by (replicated in) yeast cells. YACs can accept humongous foreign DNA inserts! This is because to be a chromosome that will replicate in a yeast cell requires one centromere and two telomeres… and little else!
Recall that telomeres are needed in replication to keep the chromosome from shortening during replication of the DNA. The centromere is needed to attach chromatids to spindle fibers so that they can separate during anaphase in mitosis (and meiosis). So along with a centromere and two telomeres, just include restriction sites to enable recombination with inserts as long as 2000 Kbp. That’s a YAC! The tough part of course is keeping a 2000Kbp long DNA fragment intact long enough to get it into the YAC.
However a vector is engineered and chosen, sequencing its insert can tell us many things. They can show us how a gene is regulated by revealing known and uncovering new regulatory DNA sequences. They can tell us what other genes are nearby, and where genes are on chromosomes. Genomic DNA sequences from one species can probe for similar sequences in other species and comparative sequence analysis can then tell us a great deal about gene evolution and the evolution of species.
One early surprise from gene sequencing studies was that we share many common genes and DNA sequences with other species, from yeast to worms to flies… and of course vertebrates and our more closely related mammal friends. You may already know that the chimpanzee’s and our genomes are 99% similar. Moreover, we have already seen comparative sequence analysis showing how proteins with different functions nevertheless share structural domains.
Let’s look at cloning a genomic library in phage. As you will see, the principles are similar to cloning a foreign DNA into a plasmid, or in fact any other vector, but the numbers and details used here exemplify cloning in phage.
A. Preparing Genomic DNA of a Specific Length for Cloning
To begin with, high molecular weight (i.e., long molecules of) the desired genomic DNA are isolated, purified and then digested with a restriction enzyme. Usually, the digest is partial, aiming to generate overlapping DNA fragments of random length. When the digest is electrophoresed on agarose gels, the DNA (stained with ethidium bromide, a fluorescent dye that binds to DNA) looks like a bright smear on the gel. All of the DNA could be recombined with suitably digested vector DNA. But, to further reduce the number of clones to be screened for a sequence of interest, early cloners would generate a Southern blot (named after Edward Southern, the inventor of the technique) to determine the size of genomic DNA fragments most likely to contain a desired gene.
Beginning with a gel of genomic DNA restriction digests, the Southern blot protocol is illustrated below
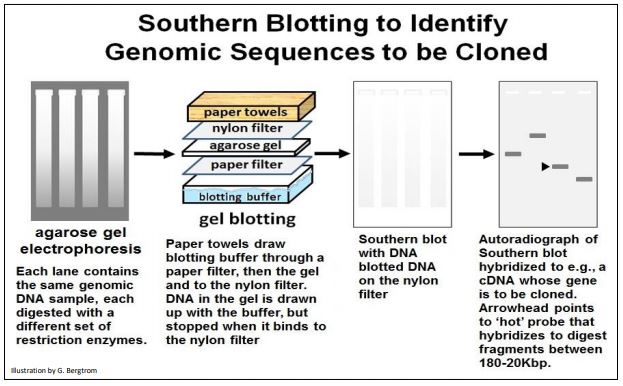
To summarize the steps:
a) Digest genomic DNA with one or more restriction endonucleases.
b) Run the digest products on an agarose gel to separate fragments by size (length). The DNA appears as a smear when stained with a fluorescent dye.
c) Place a filter on the gel. The DNA transfers (blots) to the filter for e.g., 24 hours
d) Remove the blotted filter and place it in a bag containing a solution that can denature the DNA.
e) Add radioactive probe (e.g., cDNA) containing the gene or sequence of interest. The probe hybridizes (bind) to complementary genomic sequences on the filter
f) Prepare an autoradiograph of the filter and see a ‘band’ representing the size of genomic fragments of DNA that include the sequence of interest.
Once you know the size (or size range) of restriction digest fragments that contain the DNA you want to study, you are ready to:
a) run another gel of digested genomic DNA.
b) cut out the piece of gel containing the fragments that ‘lit up’ with your probe in the autoradiograph.
c) remove (elute) the DNA from the gel piece into a suitable buffer
d) prepare the DNA for insertion into (recombination with) a genomic cloning vector
B. Recombining Size-Restricted Genomic DNA with Phage DNA
After elution of restriction digested DNA fragments of the right size range from the gels, the DNA is mixed with compatibly digested phage DNA at concentrations that favor the formation of H-bonds between the ends of the phage DNA and the genomic fragments. Addition of DNA ligase covalently links the recombined DNA molecules. These steps are abbreviated in the illustration below.

The recombinant phage that are made next will contain sequences that become the genomic library.
C. Creating Infectious Viral Particles with Recombinant Phage DNA
The next step is to package the recombined phage DNA with added purified viral coat proteins to make infectious phage particles (below)
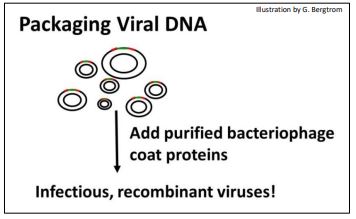
269 Genomic Libraries: Make and Package Recombinant Phage DNA
Packaged phage are added to a culture tube full of host bacteria (typically E. coli). After infection, the recombinant DNA enters the cells where it replicates and directs the production of new phage that eventually lyse the host cell (illustrated below).
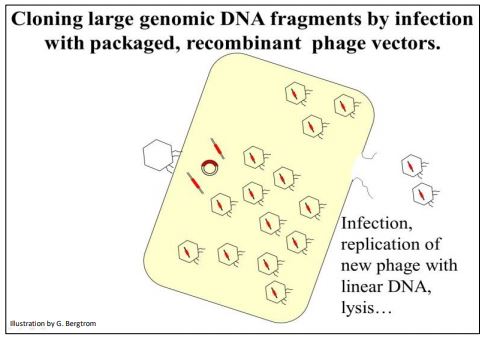
The recombined vector can also be introduced directly into the host cells by transduction (which is to phage DNA what transformation is to plasmid DNA). Whether by infection or transduction, the recombinant phage DNA ends up in host cells which produce new phage that eventually lyse the host cell. The released phages go on to infect more host cells until all cells have lysed. What remains is a tube full of lysate containing cell debris and lots of recombinant phage particles.
270 Infect Host with Recombinant Phage to Make a Genomic Library
D. A Note About Some Other Vectors
We’ve seen that phage vectors accommodate larger foreign DNA inserts than plasmid vectors, and YACs even more…, and that for larger genomes, the goal is to choose a vector able to house larger fragments of ‘foreign’ DNA so that you end up screening fewer clones. Given a large enough eukaryotic genome, it may be necessary to screen more than a hundred thousand clones in a phage-based genomic library. Apart from size-selection of genomic fragments before inserting them into a vector, selecting the appropriate vector is just as important. The table below lists commonly used vectors and the sizes of inserts they will accept.
| Vector Type | Insert Size (thousands of bases) |
| Plasmids | up to 15 |
| Phage Lambda (\(\lambda \)) | up to 25 |
| Cosmids | up to 45 |
| Bacteriophage P1 | 70 to 100 |
| P1 artificial chromosomes (PACs) | 130 to 150 |
| Bacterial artificial chromosomes (BACs) | 120 to 300 |
| Yeast artificial chromosomes (YACs) | 250 to 2000 |
Click on the links to these vectors to learn more about them. We will continue this example by screening a phage lysate genomic library for a recombinant phage with a genomic sequence of interest.
E. Screening a Genomic Library; Titering Recombinant Phage Clones
A phage lysate is titered on a bacterial lawn to determine how many virus particles are present. A bacterial lawn is made by plating so many bacteria on the agar plate that they simply grow together rather than as separate colonies. In a typical titration, a lysate might be diluted 10-fold with a suitable medium and this dilution is further diluted 10-fold… and so on. Such serial 10X dilutions are then spread over bacterial (e.g., E. coli) lawns. What happens on such a culture plate?
Let’s say that when 10 μl of one of the dilutions are spread on the bacterial lawn, they infect 500 E. coli cells on the bacterial lawn. After a day or so, there will be small clearings in the lawn called plaques…, 500 of them in this example. These are 500 tiny clear spaces on the bacterial lawn created by the lysis of first one infected cell, and then progressively more and more cells neighboring the original infected cell. Each plaque is thus a clone of a single virus, and each virus particle in a plaque contains a copy of the same recombinant phage DNA molecule (below).

If you actually counted 500 plaques on the agar plate, then there must have been 500 virus particles in the 10 μl seeded onto the lawn. And, if this plate was the fourth dilution in a 10-fold serial dilution protocol, there must have been 2000 (4 X 500) phage particles in 10 μl of the original undiluted lysate.
F. Screening a Genomic Library; Probing the Genomic Library
In order to represent a complete genomic library, it is likely that many plates of such a dilution (~500 plaques per plate) will have to be created and then screened for a plaque containing a gene of interest. But, if only size-selected fragments were inserted into the phage vectors in the first place, the plaques represent only a partial genomic library, requiring screening fewer clones to find the sequence of interest. For either kind of library, the next step is to make replica filters of the plaques. Replica plating of plaques is similar to making a replica filter bacterial colonies. While much of the phage DNA in a plaque is encased in viral proteins, there will also be DNA on the plaque replicas that were never packaged into viral particles. The filters can be treated to denature the latter DNA and then directly hybridized to a probe with a known sequence. In the early days of cloning, probes for screening a genomic library were usually an already isolated and sequenced cDNA clone, either from the same species as the genomic library, or from a cDNA library of a related species. After soaking the filters in a radioactively labeled probe, X-Ray film is placed over the filter, exposed and developed. Black spots will form where the film lay over a plaque containing genomic DNA complementary to the radioactive probe. In the example illustrated below, a globin cDNA might have been used to probe the genomic library (globin genes were among the first to be cloned!).
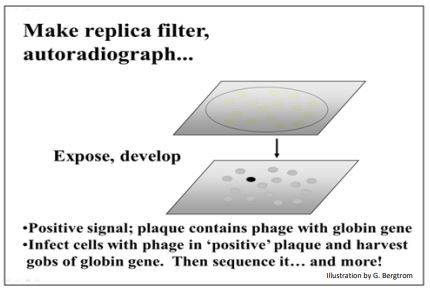
G. Isolating a Gene for Further Study
Cloned genomic DNA fragments are much longer than any gene of interest, and always longer than any cDNA from a cDNA library. They are also embedded in a genome that is thousands of times as long as the gene itself, making the selection of an appropriate vector necessary. If the genome can be screened among a reasonable number of cloned phage (~100,000 plaques for instance), the one plaque producing a positive signal on the autoradiograph would be further studied.
This plaque should contain the gene of interest. The next step is to find the gene within a genomic clone that can be as much a 20kbp long. The traditional strategy is to purify the cloned DNA, subject it to restriction endonuclease digestion, and separate of the digest particles by agarose gel electrophoresis. Using Southern Blotting, the separated DNA fragments are denatured and blotted to a nylon filter. The filter is then probed with the same tagged probe used to find the positive clone (plaque). The smallest DNA fragment containing the gene of interest can itself be subcloned in a suitable vector, and grown to provide enough DNA for further study of the gene.