
11.9: DNA Sequencing
- Page ID
- 42824

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. A Brief History of DNA Sequencing
RNA sequencing came first, when Robert Holley sequenced a tRNA in 1965. The direct sequencing of tRNAs was possible because tRNAs are small, short nucleic acids, and because many of the bases in tRNAs are chemically modified after transcription. An early method for DNA sequencing developed by Walter Gilbert and colleagues involved DNA fragmentation, sequencing of the small fragments of DNA, and then aligning the overlapping sequences of the short fragments to assemble longer sequences. Another method, the DNA synthesis-based ‘dideoxy’ DNA sequencing technique, was developed by Frederick Sanger in England. Sanger and Gilbert both won a Nobel Prize in Chemistry in 1983 for their DNA sequencing efforts. However, because of its simplicity, Sanger’s method quickly became the standard for sequencing all manner of cloned DNAs.
The first complete genome to be sequenced was that of a bacteriophage (bacterial virus) called φX174. At the same time as the advances in sequencing technology were occurring, so were some of the early developments in recombinant DNA technology. Together these led to more efficient and rapid cloning and sequencing of DNA from increasingly diverse sources. The first focus was of course on genes and genomes of important model organisms, such as E. coli, C. elegans, yeast (S. cerevisiae)…, and of course us! By 1995, Craig Venter and colleagues at the Institute for Genomic Research had completed the sequence of an entire bacterial genome (Haemophilus influenzae) and by 2001, Venter’s private group along with Frances Collins and colleagues at the NIH had published a first draft of the sequence of the human genome. Venter had proven the efficacy of a whole-genome sequencing approach called shotgun sequencing, which was much faster than the gene-by-gene, fragment-by-fragment ‘linear’ sequencing strategy being used by other investigators (more later!). Since Sanger’s dideoxynucleotide DNA sequencing method remains a common and economical methodology, let’s consider the basics of the protocol.
B. Details of DiDeoxy Sequencing
Given a template DNA (e.g., a plasmid cDNA), Sanger used in vitro replication protocols to demonstrate that he could:
- Replicate DNA under conditions that randomly stopped nucleotide addition at every possible position in growing strands.
- Separate and then detect these DNA fragments of replicated DNA.
Recall that DNA polymerases catalyze the formation of phosphodiester bonds by linking the \(\alpha \) phosphate of a nucleotide triphosphate to the free 3’ OH of a deoxynucleotide at the end of a growing DNA strand. Recall also that the ribose sugar in the deoxynucleotide precursors of replication lack a 2’ OH (hydroxyl) group. Sanger’s trick was to add dideoxynucleotide triphosphates to his in vitro replication mix. The ribose on a dideoxynucleotide triphosphate (ddNTP) lacks a 3’ OH, in addition to the 2’ OH group (as shown below).
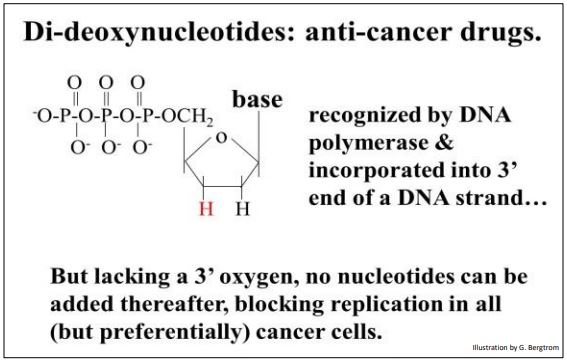
Adding a dideoxynucleotide to a growing DNA strand stops replication. No further nucleotides can add to the 3’-end of the replicating DNA strand because the 3’–OH necessary for the dehydration synthesis of the next phosphodiester bond is absent! Because they can stop replication in actively growing cells, ddNTPs such as dideoxyadenosine (tradename, cordycepin) are anti-cancer chemotherapeutic drugs.
266 Treating Cancer with Dideoxynucleosides
A look at a manual DNA sequencing protocol reveals what is going on in the sequencing reactions. Four reaction tubes are set up, each containing the template DNA to be sequenced, a primer of known sequence and the four required deoxynucleotide precursors necessary for replication.
The set-up for manual DNA sequencing is shown below.
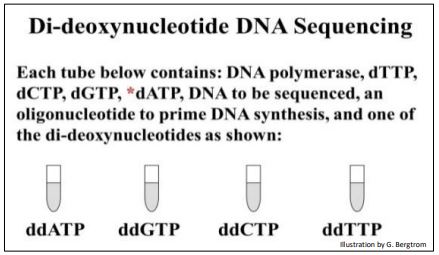
A different ddNTP, (ddATP, ddCTP, ddGTP or ddTTP) is added to each of the four tubes. Finally, DNA polymerase is added to each tube to start the DNA synthesis reaction. During DNA synthesis, different length fragments of new DNA accumulate as the ddNTPs incorporate randomly, opposite complementary bases in the template DNA being sequenced. The expectations of the didieoxy sequencing reactions in the four tubes are illustrated below.
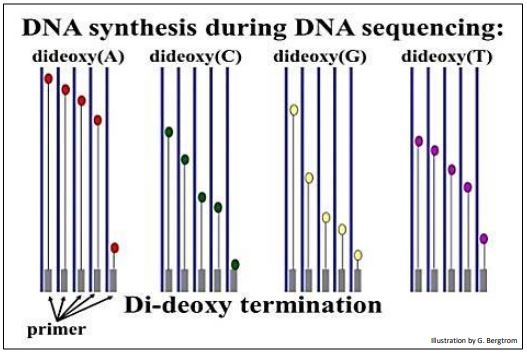
A short time after adding the DNA polymerase to begin the reactions, the mixture is heated to separate the DNA strands and fresh DNA polymerase is added to repeat the synthesis reactions. These sequencing reactions are repeated as many as 30 times in order to produce enough radioactive DNA fragments to be detected. When the heat-stable Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus became available ( more later!), it was no longer necessary to add fresh DNA polymerase after each replication cycle. The many heating and cooling cycles required for what became known as chain-termination DNA sequencing were soon automated using inexpensive programmable thermocyclers.
Since a small amount of a radioactive deoxynucleotide (usually 32P-labeled ATP) was present in each reaction tube, the newly made DNA fragments are radioactive. After electrophoresis to separate the new DNA fragments in each tube, autoradiography of the electrophoretic gel reveals the position of each terminated fragment. The DNA sequence can then be read from the gel as illustrated in the simulated autoradiograph below.
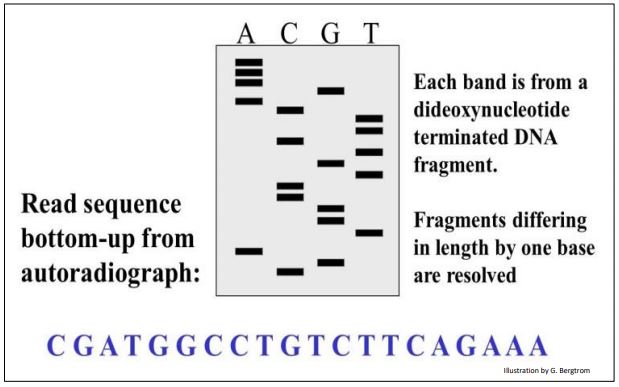
As shown in the cartoon, the DNA sequence can be read by reading the bases from the bottom of the gel, starting with the C at the bottom of the C lane. Try reading the sequence yourself!
The first semi-automated DNA sequencing method was invented in Leroy Hood’s California lab in 1986. Though still Sanger sequencing, the four dideoxynucleotides in the sequencing reaction were tagged for detection with a fluorescent dyes instead radioactive phosphate-tagged nucleotides. After the sequencing reactions, the reaction products are electrophoresed on an ‘automated DNA sequencer’. UV light excites the migrating dye-terminated DNA fragments as they pass through a detector. The color of their fluorescence is detected, processed and sent to a computer, generating color-coded graph like the one below, showing the order (and therefore length) of fragments passing the detector and thus, the sequence of the strand.
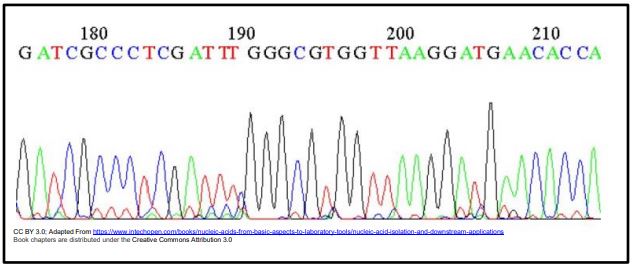
A most useful feature of this sequencing method is that a template DNA could be sequenced in a single tube, containing all the required components, including all four dideoxynucleotides! That’s because the fluorescence detector in the sequencing machine separately sees all the short ddNTP-terminated fragments as they move through the electrophoretic gel.
Hood’s innovations were quickly commercialized making major sequencing projects possible, including whole genome sequencing. The rapidity of automated DNA sequencing led to the creation of large sequence databases in the U.S. and Europe.
The NCBI (National Center for Biological Information) maintains the U.S. database. Despite its location, the NCBI archives virtually all DNA sequences determined worldwide. New ‘tiny’ DNA sequencers have made sequencing DNA so portable that in 2016, one was even used in the International Space Station. Expanding databases and new tools and protocols (some are described below) to find, compare and analyze DNA sequences have also grown rapidly.
C. Large Scale Sequencing
Large-scale sequencing targets entire prokaryotic, and typically much larger eukaryotic genomes. The latter require strategies that either sequence long DNA fragments and/or sequencing DNA fragments more quickly. We already noted the shotgun sequencing used by Venter to sequence smaller and larger genomes (including our own… or more accurately, his own!). In shotgun sequencing, cloned DNA fragments 1000 base pairs or longer are broken down at random into smaller, more easily sequenced fragments. The fragments are themselves cloned and sequenced and non-redundant sequences are assembled by aligning overlapping regions of sequence. Today’s computer software is quite adept at rapid overlapping sequence alignment as well as connecting and displaying long contiguous DNA sequences. Shotgun sequencing is summarized below.

Sequence gaps that remain after shotgun sequencing can be filled in by primer walking, in which a known sequence near the gap is the basis of creating a sequencing primer to “walk” into the gap region on an intact DNA that has not been fragmented. Another ‘gap-filling’ technique involves PCR (the Polymerase Chain Reaction, to be described shortly). Briefly, two oligonucleotides are synthesized based on sequence information on either side of a gap. Then PCR is used to synthesize the missing fragment, and the fragment is sequenced to fill in the gap.