
3.8: Lysosomes and Peroxisomes
- Page ID
- 3972

A cell is composed of many different organelles and microbodies (or cytosomes) is a type of organelle that is found in the cells of plants, protozoa, and animals. Organelles in the microbody family include peroxisomes, glyoxysomes, glycosomes and hydrogenosomes.
Lysosomes
Lysosomes are roughly spherical bodies enclosed by a single membrane. They are manufactured by the Golgi apparatus (Figure \(\PageIndex{1}\)) and contain over 50 different kinds of hydrolytic enzymes including proteases, lipases, nucleases, and polysaccharidases. The pH within the lysosome is about pH 5, substantially less than that of the cytosol (~pH 7.2). All the enzymes in the lysosome work best at an acid pH, which reduces the risk of their digesting their own cell if they should escape from the lysosome.
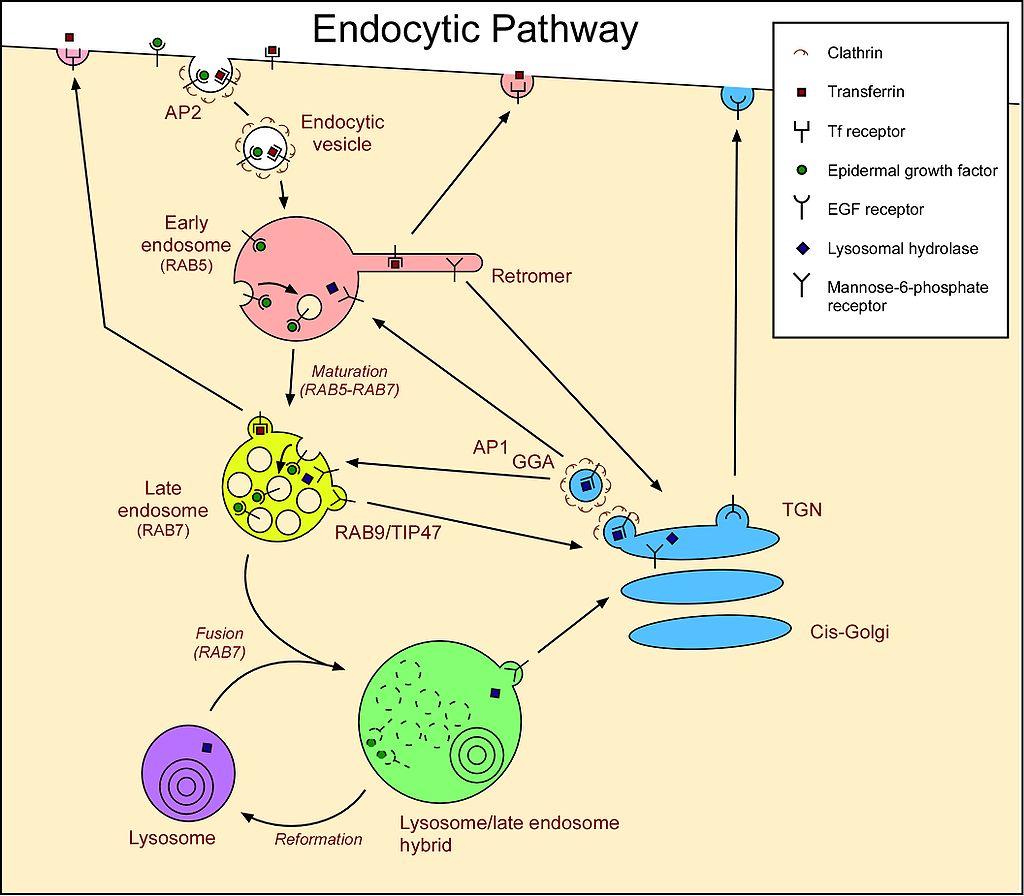
Materials within the cell scheduled for digestion are first deposited within lysosomes. These may be:
- other organelles, such as mitochondria, that have ceased functioning properly and have been engulfed in autophagosomes
- food molecules or, in some cases, food particles taken into the cell by endocytosis
- foreign particles like bacteria that are engulfed by neutrophils
- antigens that are taken up by
- "professional" antigen-presenting cells like dendritic cells (by phagocytosis) and
- B cells (by binding to their antigen receptors (BCRs) followed by receptor-mediated endocytosis.
At one time, it was thought that lysosomes were responsible for killing cells scheduled to be removed from a tissue; for example, the resorption of its tail as the tadpole metamorphoses into a frog. This is incorrect. These examples of programmed cell death (PCD) or apoptosis take place by an entirely different mechanism.
In some cells, lysosomes have a secretory function — releasing their contents by exocytosis.
- Cytotoxic T cells (CTL) secrete perforin from lysosomes.
- Mast cells secrete some of their many mediators of inflammation from modified lysosomes.
- Melanocytes secrete melanin from modified lysosomes.
- The exocytosis of lysosomes provides the additional membrane needed to quickly seal wounds in the plasma membrane.
Lysosomal storage diseases are caused by the accumulation of macromolecules (proteins, polysaccharides, lipids) in the lysosomes because of a genetic failure to manufacture an enzyme needed for their breakdown. Neurons of the central nervous system are particularly susceptible to damage. Most of these diseases are caused by the inheritance of two defective alleles of the gene encoding one of the hydrolytic enzymes. Examples include:
- Tay-Sachs disease and Gaucher's disease — both caused by a failure to produce an enzyme needed to break down sphingolipids (fatty acid derivatives found in all cell membranes).
- Mucopolysaccharidosis I (MPS-I). Caused by a failure to synthesize an enzyme (α-L-iduronidase) needed to break down proteoglycans like heparan sulfate. In April 2003, the U.S. Food and Drug Administration approved a synthetic version of the enzyme, laronidase (Aldurazyme®), as a possible treatment. This enzyme (containing 628 amino acids) is manufactured by recombinant DNA technology.
However, one lysosomal storage disease, I-cell disease ("inclusion-cell disease"), is caused by a failure to "tag" (by phosphorylation) all the hydrolytic enzymes that are supposed to be transported from the Golgi apparatus to the lysosomes. Lacking the mannose 6-phosphate (M6P) tag, they are secreted from the cell instead. The result: all the macromolecules incorporated in lysosomes remain undegraded forming "inclusion bodies" in the cell.
Peroxisomes
Peroxisomes, also called microbodies, are about the size of lysosomes (0.5–1.5 µm) and like them are enclosed by a single membrane. They also resemble lysosomes in being filled with enzymes. However, peroxisomes bud off from the endoplasmic reticulum, not the Golgi apparatus (the source of lysosomes) and the enzymes and other proteins destined for peroxisomes are synthesized in the cytosol. Each contains a peroxisomal targeting signal (PTS) that binds to a receptor molecule that takes the protein into the peroxisome and then returns for another load. Two peroxisomal targeting signals have been identified: a 9-amino acid sequence at the N-terminal of the protein and a tripeptide at the C-terminal. Each has its own receptor to take it to the peroxisome.
Functions of the peroxisomes in the human liver inlcude:
- Breakdown (by oxidation) of excess fatty acids.
- Breakdown of hydrogen peroxide (H2O2), a potentially dangerous product of fatty-acid oxidation. It is catalyzed by the enzyme catalase.
- Participates in the synthesis of cholesterol. One of the enzymes involved, HMG-CoA reductase, is the target of the popular cholesterol-lowering "statins".
- Participates in the synthesis of bile acids.
- Participates in the synthesis of the lipids used to make myelin.
- Breakdown of excess purines (AMP, GMP) to uric acid.
Peroxisomes are also present in plant cells where they participate is such functions as symbiotic nitrogen fixation and photorespiration.
A variety of rare inherited disorders of peroxisome function occur in humans. Most involve mutant versions of one or another of the enzymes found within peroxisomes. For example: X-linked adrenoleukodystrophy (X-ALD) results from a failure to metabolize fatty acids properly. One result is deterioration of the myelin sheaths of neurons. The disorder occurs in young boys because the gene is X-linked. An attempt to find an effective treatment was the subject of the 1992 film Lorenzo's Oil. A few diseases result from failure to produce functional peroxisomes. For example: Zellweger syndrome results from the inheritance of two mutant genes for one of the receptors (PXR1) needed to import proteins into the peroxisome.