
9.2: Eukaryotic Transcriptional Regulation
- Page ID
- 16143

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)As with almost every comparison with prokaryotic systems, regulation of eukaryotic transcription is much more complex than prokaryotic gene control, although still based on similar mechanisms of activators and repressors. There is no close eukaryotic equivalent to operons, though: eukaryotic genes are always transcribed one per mRNA. The previous chapter described the formation of a preinitiation complex of transcription factors for RNA polymerase II. These transcription factors (e.g. TFIID, TFIIH, etc.) are known as general transcription factors, and are required for transcription of any gene at any level. However, there are also specific transcription factors, usually referred to simply as transcription factors (TF), that modulate the frequency of transcription of particular genes. Some upstream elements and their associated TFs are fairly common, while others are gene or gene-family specific. An example of the former is the upstream element AACCAAT and its associated transcription factor, CP1. Another transcription factor, Sp1, is similarly common, and binds to a consensus sequence of ACGCCC. Both are used in the control of the beta-globin gene, along with more specific transcription factors, such as GATA-1, which binds a consensus AAGTATCACT and is primarily produced in blood cells. This illustrates another option found in eukaryotic control that is not found in prokaryotes: tissue-specific gene expression. Genes, being in the DNA, are technically available to any and every cell, but obviously the needs of a blood cell differ a great deal from the needs of a liver cell, or a neuron. Therefore, each cell may produce transcription factors that are specific to its cell or tissue type. These transcription factors can then allow or repress expression of multiple genes that help de ne this particular cell type, assuming they all have the recognition sequences for the TFs. These recognition sequences are also known as response elements (RE).
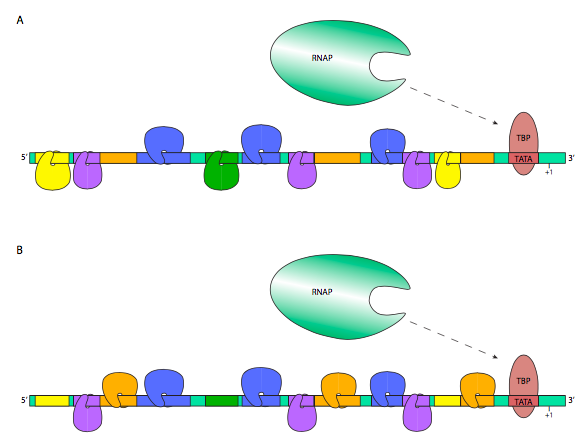
Very often, a combination of many transcription factors, both enhancers and silencers, is responsible for the ultimate expression rate of a given eukaryotic gene. This can be done in a graded fashion, in which expression becomes stronger or weaker as more enhancers or silencers are bound, respectively, or it can be a binary mode of control, in which a well-defined group of TFs are required to turn on transcription, and missing just one can effectively shut down transcription entirely. In the first case, activating TFs generally bind to the GTFs or RNA Polymerase II directly to help them recognize the promoter more efficiently or stably, while repressing TFs may bind to the activating TFs, or to the GTFs or RNAP II, in preventing recognition of the promoter, or destabilizing the RNAP II preinitiation complex. In the second case, activation hinges on the building of an enhanceosome, in which transcription factors and protein scaffolding elements and coactivators come together to position and stabilize the preinitiation complex and RNAP II on the promoter. The most prominent and nearly ubiquitous coactivator is named Mediator, and binds to the CTD of the β’ subunit of RNA polymerase II and also to a variety of transcription factors.

Eukaryotic transcription factors, while varied, usually contain at least one of the following transcription factor motifs: zinc fingers, leucine zippers, basic helix-loop-helix domains, Rel homology region domains, or a variation thereof.
The zinc finger motif was the first DNA-binding domain to be discovered, and was found in a general transcription factor associated with RNA polymerase III. The initial structure found was a repeating ~30-amino acid motif with two invariant Cys and two invariant His residues that together bind a Zn2+ ion and thus bring a tight loop or “ finger” of basic potentially DNA-binding residues together. The basic finger binds to the major groove of the DNA, with the exact sequence-matching characteristics determined by the topology of the particular residues that make up the finger. Although most DNA binding motifs insert a positively-charged α-helical domain into the major groove of DNA, the zinc-finger proteins are the only ones that combine several such motifs to interact with the DNA in several sequential sites.
In addition to the first type of Zn2+-binding site described with two Cys and two His (Cys2-His2), there are two major variations to note. The first is the Cys2-Cys2 type, which is characteristic of steroid receptor transcription factors such as the glucocorticoid receptor or estrogen receptor. We will consider them in more detail later with the discussion of intracellular signal transduction, but for now, the general idea is that unactivated steroid hormone receptors are found in the cytoplasm, where they come in contact with and bind their cognate hormone molecule. They then translocate to the nucleus, where they dimerize and are able to act as transcription factors. The second major variation of the zinc finger is the binuclear Cys6, which carries six Cys residues to create a slightly larger “basket” in which two Zn2+ ions are held, rather than just one. The best-studied example of this type of zinc-finger protein is GAL4, a yeast metabolic transcription factor.

The next motif is the leucine zipper. Although this is a common motif for transcription factors, it is important to note that unlike the zinc-finger, the leucine zipper itself is not a DNA-binding motif. Rather, it is a protein dimerization motif, and determines the way in which two protein subunits interact. However, the leucine zipper is a common structural motif in transcription factors. It works through opposing domains of regularly spaced hydrophobic amino acids, particularly leucines, which are very effective at holding the two subunits together in the aqueous environment of the cell. The leucines are found in every 7th residue position of an α-helical domain, leading to a coiled-coil superstructure when two subunits interact. The (+) charged DNA-binding domains of these proteins are usually N-terminal to the leucine zippers, as in the case of the bZIP category of leucine zipper proteins (the name stands for basic region leucine zipper).

The bHLH, or basic helix-loop-helix domains appear to be elaborations on the leucine zipper theme. In this case, the N-terminal region is highly basic, making it ideal for interacting with DNA, and this basic domain, which is also helical, leads into the first helix (H1) of the motif, which is then connected by a non-helical loop of amino acids, leading into a second helical region (H2). Beyond the bHLH, these transcription factors may merge into a leucine zipper motif or other protein interaction domain for dimerization. Though the primary binding domain is N-terminal to H1, the H1 domains also appear to play a role in binding the major groove of the DNA. [Example myc]

NF-kB (nuclear factor kB) is a ubiquitous transcription factor discovered (and most noticeable) in the immune system. When active, it is a heterodimer, with both subunits containing a Rel homology region (RHR). Rel is an oncogene, and the RHR are named for their similarity to the previously-sequenced rel. The RHR domains bind to DNA with extraordinary affinity, due in part to having ve loops for DNA contact per subunit. Just as with the other types of transcription factors, some RHR-containing proteins are repressors, while others are activators.
The regulation of NF-kB is rather interesting: once it is in the nucleus, it is generally active. However, it is, as almost all cellular proteins, made in the cytoplasm. Inhibitors of NF-kB (IkB) also reside in the cytoplasm, and they act by binding the NF-kB and covering the nuclear localization signal that allows its import into the nucleus. Thus sequestered, the NF-kB must remain in the cytoplasm inactive until some stimulus activates IkB kinase, which phosphorylates the IkB and leads to ubiquitination and degradation, finally releasing the NF-kB from its bonds.
Because it can be mobilized quickly (compared to synthesizing new protein), NF-kB is consider a rapid-response transcription factor that is often used to begin expression of a gene needed soon after it has been “ordered” by a signal, either extracellular or intracellular. Not surprisingly for a factor discovered in the immune system, it is activated in response to bacterial and viral antigens, as well as other types of cellular stress or insult.

In addition to the relatively short-term regulation of gene expression controlled by binding transcription factors to regulatory elements, there are also stronger methods of locking away a gene to prevent its expression. In chapter 7, acetylation and deacetylation of histones was discussed as a method for decreasing and increasing their affinity for DNA. This can be controlled (Figure \(\PageIndex{11}\)B) by the recruitment of histone deacetylase (HDAC) to particular genes via repressor/co-repressor complexes. The deacetylase forces tight winding of the targeted DNA to the histones, precluding access by RNA polymerases or general transcription factors.

Another recruiter of HDAC are MBD proteins, which bind to methylated DNA. DNA methylation in mammals usually occurs on CpG dinucleotide sequences. This methylation appears to have the effect of blocking access of transcription factors and enzymes to the DNA. It can do so directly, or by recruiting MBD (methyl-CpG-binding domain) proteins. In either case, methylation is a long-term method of locking up genes, and is the mechanism for turning off genes that would never be used in a particular cell type (e.g. hemoglobin in neurons).