
8.9: DNA Miniprep by Alkaline Lysis (Activity)
- Page ID
- 24784

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Alkaline Lysis
Once DNA is introduced and carried in bacteria, we would like to isolate the DNA again for further manipulation. In order to do so, bacteria containing the plasmid of interest is grown in a liquid culture of nutrient-rich broth made of yeast extract called Luria-Bertani Broth (LB). These cultured bacteria are grown until they are of a high concentration overnight. They are harvested through centrifugation and the broth is removed. The resulting pellet of bacteria is resuspended in a physiological buffer containing the chelator EDTA. A chelator is a chemical that removes divalent cations like Ca2+ or Mg2+ from solution. This is significant because divalent cations are necessary for DNA digesting enzymes to be active. By chelating the ions, the DNA we ultimately wish to purify will be safe from degradation.
After resuspension of the bacteria, an alkaline solution of 0.1N NaOH is mixed into the bacterial mix. This solution also contains an ionic detergent called sodium dodecyl sulfate (SDS) that aids in denaturing proteins and disrupting their interactions with the DNA. The mixture becomes viscous as the bacteria burst open and their contents leak into the solution. This basic solution is then neutralized with a potassium acetate buffer at pH5.5. As the solutions mix together, the pH approaches 7 and the potassium interacts with the SDS to cause the precipitation of the genomic chromosomal DNA and proteins. In order to separate the precipitate from the solution, the mixture is centrifuged at high speed to pellet the genomic DNA and protein. The supernatant, or solution, is transferred to a column containing a silica membrane. Under high salt conditions, DNA adheres to glass or silica. By passing the solution through this column, the plasmid DNA in the supernatant is trapped onto the silica membrane and removed from the solution. Additional washes are used to removed stray contaminants and remove the excessive salt. Plasmid DNA is finally removed from the column through elution by a low salt buffer. This low salt buffer is Tris pH 8 with EDTA (TE). Plasmid DNA can be stored stably in the TE buffer in the freezer for extended periods.
Exercise 1: Plasmid DNA Mini-Prep by Alkaline Lysis
- Inoculate 2 ml of rich medium (LB, YT, or Terrific Broth) containing the appropriate antibiotic with a single colony of transformed bacteria. Incubate the culture overnight at 37°C with vigorous shaking. (This is what you were provided)
- Each group should take 2 cultures
- Centrifuge culture tubes directly at maximum for 5 minutes.
- If incapable of spinning in these tubes, transfer 1.5 ml of the culture into a microfuge tube (Eppendorf Tube).
- Centrifuge at maximum speed for 30 sec.
- When centrifugation is complete, pour the broth solution into a container of bleach.
- Resuspend the bacterial pellet in 250 μl of ice-cold P1 solution by vigorous shaking and transfer back into a microcentrifuge tube.
- P1 is a physiological solution of 50mM Tris at pH 8.
- P1 contains a Chelator called EDTA.
- Chelators bind up excess divalent cations that are required for DNAse activity.
- Lyse: Add 250μl of P2 solution to each bacterial suspension. Close the tube tightly, and mix the contents by inverting the tube gently five times. Do not vortex! Store the tube on ice.
- This is the lysis buffer containing the detergent Sodium Dodecyl Sulfate and NaOH.
- Neutralize: Add 350 μl of ice-cold P3 solution. Close the tube and disperse lysis solution by inverting the tube several times. Store the tube on ice for 3-5 minutes.
- This is the neutralization buffer containing Potassium Acetate.
- Neutralization restores pH to near 7 and also causes the precipitation of genomic DNA and proteins into a gloopy mess (snot-like).
- Centrifuge the bacterial lysate at maximum speed for 5 minutes in a microfuge.
- Snot-like substances should be tightly packed into a pellet at the bottom of the tube after this step.
- The solution or supernatant contains the plasmid DNA.
- Column Purification of DNA: Transfer the supernatant to a fresh tube with a silica-membrane column
- DNA likes to bind to glass under high salt conditions.
- The white membrane is made of glass fiber.

- Centrifuge the supernatant through the column for 1 minute at maximum speed in a microfuge.
- The DNA will be bound to the membrane on the column (silica)
- Wash: Discard flow-through and place column back into the waste tube. Wash column with 500 μl PE. Centrifuge the supernatant through the column for 1 minute at maximum in a microfuge.
- PE is a solution that helps to wash away the non-specifically bound substances
- Discard flow-through and place column back into the waste tube. Wash Column with 700 μl PE. Centrifuge the supernatant through the column for 1 minute at maximum in a microfuge. Discard flow-through and repeat spin to dry column.
- Place column in a fresh centrifuge tube and Elute the nucleic acids in 50 μl of TE (pH 8.0) by binding for 1 minute and spinning at maximum speed for 1 minute.
Identification of Plasmid DNA
Once plasmids are isolated, they require identification. Plasmid vectors have known sequences and are mapped of their major features. Knowing the sequence of these pieces of DNA means knowing the locations of RE digestion sites. By using Res, digesting plasmids into known sizes aids in the verification of plasmid identity without the need to have the entire plasmid re-sequenced. A common plasmid is called pUC18 or pUC19. The “p” stands for plasmid, the “UC” stands for University of California (where it was designed) and 18 or 19 refer to the difference in the MCS. This plasmid is 2,686 base pairs or ~2.7kb (kilobase) long with a single EcoRI site in the MCS. Another plasmid of interest in learning Molecular Biology is called pGlo. This plasmid has a jellyfish gene in the MCS that codes for a protein that will fluoresce green when expressed under UV light. pGlo is 5.4kb long and contains a single EcoRI site. Once digested, by an enzyme, these plasmids can be identified based on size separation on an agarose gel. Usually, it is best to identify by using 2 different REs. Digestion is important before size comparisons since circular DNA migrates through agarose differently than linear DNA. Additionally, circular DNA can sometimes be “super-coiled” and lead to very rapid migration despite the size.
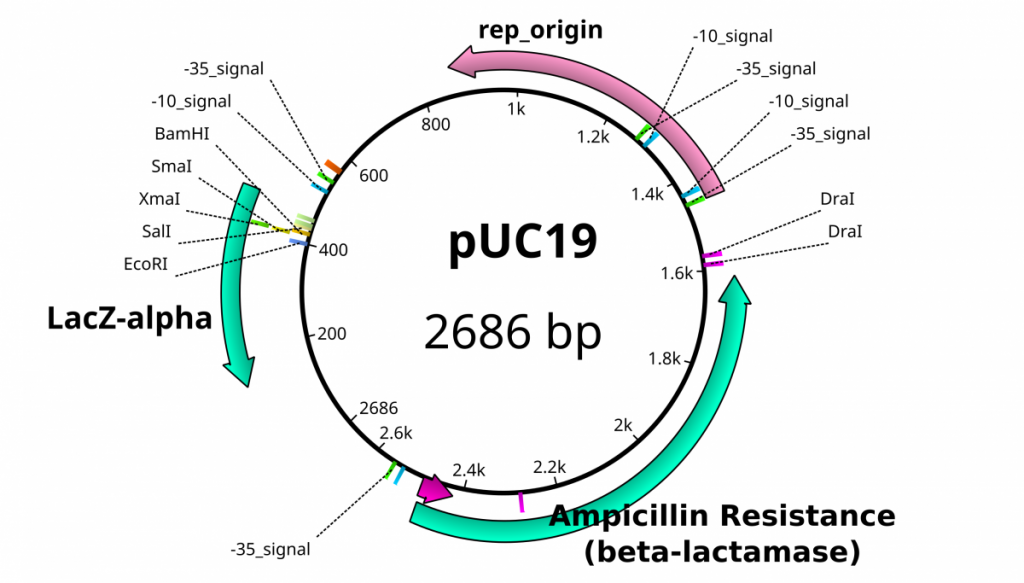
Feature map of pUC19 including the locations of some restriction enzymes.
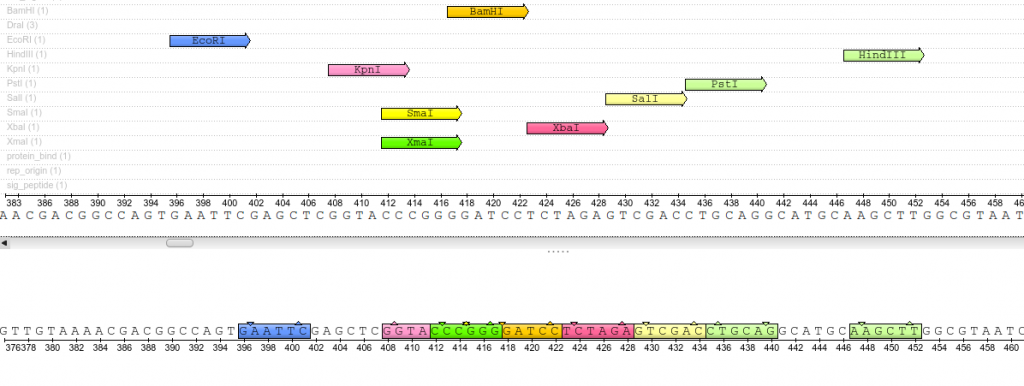
Close up view of the pUC19 Multiple Cloning Site (MCS). These restriction sites appear only once throughout the entirety of the plasmid sequence.
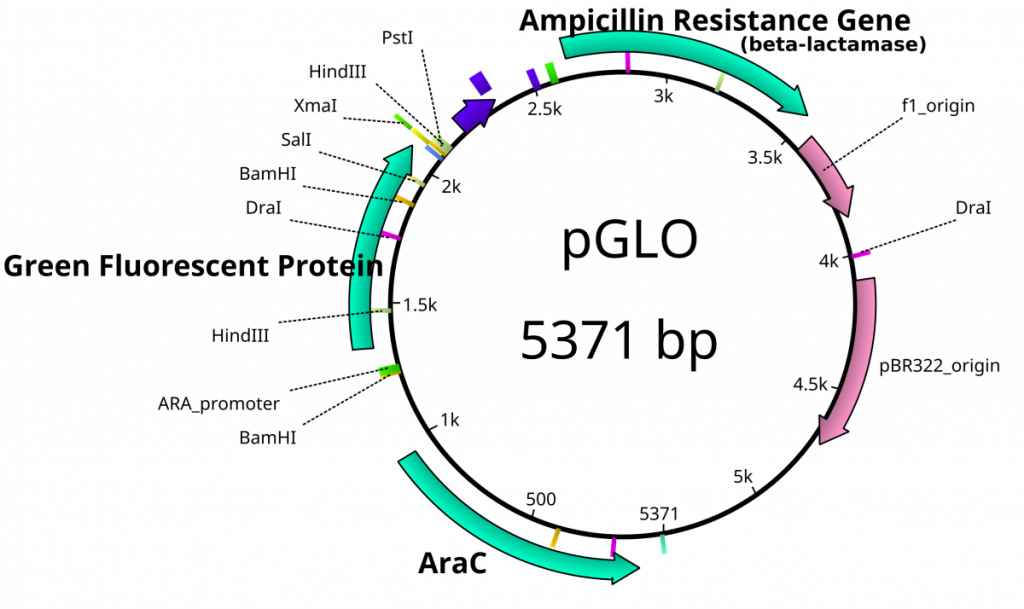
Plasmid feature map of pGlo. This is not a cloning vector, so an MCS does not exist.
Exercise 2: Restriction Digestion Identification of Plasmids
- The class should prepare 2X 0.8% agarose gels by preparing 0.4g agarose in 50ml TBE buffer.
- Melt agarose solution by microwaving for 1 minute.
- Add 5μl Sybr Safe solution into the 100ml gel solution.
- Pour this solution into a casting tray inside the refrigerator.
- Insert a comb.
- To a new tube add 2μl of plasmid DNA to 8μl of EcoRI fast digest mixture.
- 1μl Fast Digest Buffer
- 1μl Fast Digest EcoRI enzyme
- 6μl H2O
- Incubate at 37°C for 10 minutes.
- Add 2μl of loading buffer to digestion mixture.
- In a separate tube, combine 3μl plasmid DNA with 2μl loading buffer and 7μl of H2O.
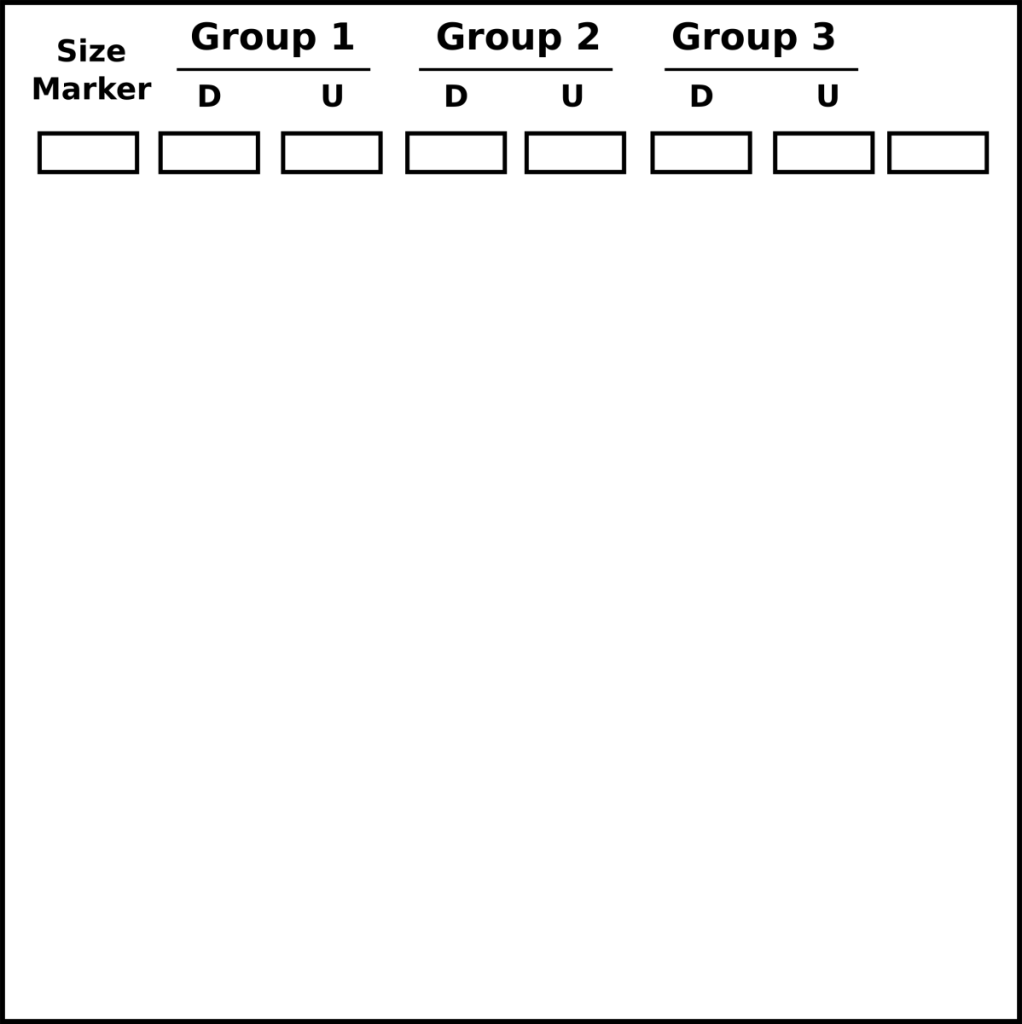
- Load gel with an appropriate size ladder in the first lane, load the Digested plasmid in the next lanes, then the undigested plasmid.
- 3 groups can load onto one gel
- D=digested plasmid
- U=undigested plasmid
- Run the gel at 110V for 30 minutes and visualize on a UV transilluminator.
- Document with your camera.
Additional Resources
- For help on this problem, please try out the In silico digestion activity.