
5.4: Chromatography
- Page ID
- 18150

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)"Chroma" refers to color and "graphy" refers to writing.
Chromatography involves the physical separation of a mixture of compounds, where historically the identification of the individual compounds is by their unique color. Chromatography can be used as a purification method (preparative scale) and also for the identification of compounds based on their chromatographic behavior (analytical scale).
There are many types of chromatography, but all involve dissolving the compounds to be separated in a liquid solution (the "mobile" phase) and then passing this solution across (or through) some solid support (or "matrix") known as the "stationary" phase. The stationary phase is typically a slurry of solid beads of silica or polysaccharide compounds, or a solid surface of these compounds. As the mobile phase travels through or across the solid phase the dissolved solutes interact to varying degrees with the stationary phase (i.e non-covalent attractive forces). The stronger the attraction, the more the molecules are retarded in comparison to the mobile phase. If there is no attraction for the solid support, then the solute molecules move with the mobile phase. The different rates of movement for the different solutes results in their separation from each other.
An example you can do at home involves a coffee filter (stationary phase) and water (mobile phase) in the separation and analysis of the dye in a water soluble marker pen (the mixture of compounds). If you draw a line with a purple marker on a strip of the filter paper, and place the bottom in a dish of water, the water will wick up the paper, and the differential affinity of the dye inks for the solid support (cellulose) results in differential migration. In the example below, the red ink has some affinity for the paper, but the blue ink does not (and migrates with the front edge of the mobile phase).
.png?revision=1&size=bestfit&width=123&height=288)
Figure 5.4.1: Chromatography of purple pen
Chromatography in biochemistry typically utilizes not paper, but beads of polysaccharide (often chemically derivatized) packed into a column, as the solid support. The solid support is often called the chromatography "resin". Some common types of chromatographic resins include:
- Ion exchange
- Affinity
- Hydrophobic
- Gel filtration
Often, the solutes to be separated are proteins, and so the discussion will focus upon protein chromatography (but the principles are the same for any solute)
Ion exchange
Ion exchange resins contain charged groups.
- These may be acidic in nature (in which case the resin is a cation exchanger)
- or basic (in which case it is an anion exchanger).
- Cation and anion exchangers may be broken down further into weak and strong exchangers (reflecting binding affinity).
|
Type of exchanger |
Functional group |
Common name |
|
Weak cation exchanger |
carboxymethyl (-) |
CM cellulose/sephadex |
|
Strong cation exchanger |
sulfopropyl (-) |
SP sephadex |
|
Weak anion exchanger |
diethylaminoethyl (+) |
DE cellulose/sephadex |
|
Strong anion exchanger |
quaternary amine (+) |
QAE sephadex |
Usually, samples are loaded under low ionic strength conditions (which promotes electrostatic interactions) and bound material is eluted using either a step or gradient elution of buffer with higher ionic strength.
- Generally speaking, a protein will bind to a cation exchange resin if the buffer pH is lower than the isoelectric point (pI) of the protein, and will bind to an anion exchange resin if the pH is higher than the pI.
- Knowledge of the pI of the protein is therefore helpful in designing a purification protocol using ion exchange resins (however, you can always simply try different resins to see which works best).
Elution of proteins from ion exchange resins
Proteins bound to ion exchange resins are bound via non-covalent ionic (salt-bridge) interactions. We can compete for these ionic binding sites on the resin with other ionic groups, namely, salts
- There are two general types of methods when eluting with a salt solution: 1. Gradient elution and 2. Step elution
- A gradient elution refers to a smooth transition of salt concentration (from low to high) in the elution buffer. Weakly binding proteins elute first, and stronger binding proteins elute last (i.e. they require higher salt concentrations in the buffer to compete them off the column)
- A gradient salt concentration can be made using a gradient maker. In its simplest form, this consists of two containers (must be the same shape) connected by a siphon (or tube at the bottom). One container contains the low salt buffer, and the other contains high salt buffer. The buffer is withdrawn from the low salt container:
.png?revision=1&size=bestfit&width=433&height=376)
Figure 5.4.2: Gradient maker
- This will produce a linear gradient from low to high salt concentrations over the total volume of the gradient
- If we know the concentration range of salt over which a protein of interest will elute we can simply elute with a buffer containing that concentration of salt. This is known as a step elution.
- Step elutions are generally faster to run, and elute the protein in a smaller overall volume than with gradient elutions. They generally work best when contaminants elute at a significantly different salt concentration than the protein of interest
Note that after ion exchange chromatography the protein of interest will be in a buffer with a potentially high salt concentration. This must be taken into account before proceeding with the next step in the purification scheme
Affinity chromatography
Affinity chromatography is a general term which applies to a wide range of chromatographic media. It can be basically thought of as some inert resin to which has been attached some compound which has a specific affinity for your protein of interest.
- Thus, a specific antibody attached to an inert resin would be a type of affinity chromatography.
- Other examples might include: a protease inhibitor attached to some matrix, designed to bind a specific protease
- a cofactor bound to some matrix, designed to bind to a particular enzyme
- a metal ion bound to a matrix, designed to chelate a protein with a metal binding site, and so on.
In each case, the type of resins used and the method of attachment may vary, as will the method of elution. One generalization regarding method of elution is that the bound ligand can be competed off of the column's functional group by including in the elution buffer a high concentration of the free functional group. For example, if the functional group of the column is a cofactor, then the bound protein can be competed off the column by passing a buffer containing a high concentration of cofactor (or cofactor analog) through the column.
Other methods of elution include changing the buffer conditions such that the protein is no longer in the native state (since it is the native state which confers the structure required for the specific binding interaction). This can be achieved by changing pH or by adding denaturing agents such as urea or guanidine.
With affinity chromatography, typically the purification achieved in a single step can be dramatic - on the order of several thousand fold. Single step purifications with specific affinity columns are not unheard - in fact it is an ideal goal of purification - a matrix which recognizes only the protein of interest and none other.
Hydrophobic resins
Hydrophobic resins contain a non-polar functional group, such as an alkane or aromatic group.
- Many proteins are able to sequester such groups on their surface and this exclusion from solvent provides the basis of the binding energy (i.e. the "hydrophobic effect").
- This interaction is enhanced by increasing ionic strength, such that proteins may bind under high salt conditions and elute under low salt conditions.
- As such these columns may be used to not only provide purification, but to desalt samples (for example after an initial ammonium sulfate precipitation).
- It is usually not possible to predict in advance which particular resin will bind a given protein, this is usually determined empirically. However, the longer the alkane, or the larger the aromatic compound, the stronger the binding typically will be.
Due to the nature of hydrophobic interactions and ionic strength, hydrophobic chromatography and ion exchange chromatography can be conveniently used sequentially. For example, after ion exchange the protein is in high salt conditions, thus it can be loaded directly onto a hydrophobic column. Conversely, a hydrophobic column is eluted in low salt, which is a requirement for binding to an ion exchange resin.
A distinction should be noted between hydrophobic interaction chromatography and reverse phase chromatography
- Hydrophobic interaction chromatography is performed in aqueous solvent conditions and changes in ionic strength are used to elute the column. The protein typically binds in the native state via hydrophobic groups located on the surface of the protein. The native state is retained during the elution conditions
- Reverse phase chromatography utilizes a hydrophobic solvent (typically acetonitrile) and the binding of a ligand is a function of the phase partition between the hydrophobic nature of the solvent and column functional group. Proteins are typically denatured in such solvents and bind due to the hydrophobic nature of the entire polypeptide sequence. Since the majority of hydrophobic groups are located in the core of globular proteins, the binding is related to the denaturation of the protein and the accessibility of these groups to the column functional groups. Proteins can be purified using reverse phase chromatography, but usually must be refolded in some way to regain functionality (i.e. the native state)
Gel filtration
Gel filtration does not rely on any chemical interaction with the protein, rather it is based on a physical property of the protein - that being the effective molecular radius (which relates to mass for most typical globular proteins).
- Gel filtration resin can be thought of as beads which contain pores of a defined size range.
- Large proteins which cannot enter these pores pass around the outside of the beads. Therefore, the volume of the column appears smaller to a large molecule.
- Smaller proteins which can enter the pores of the beads have a larger volume that they can explore, thus the volume of the column appear larger to a small molecule.
- Both large and small molecules experience the same flow rate of mobile phase (i.e. L/min).Thus, a sample of proteins passing through a gel filtration column will separate based on molecular size: the big ones will elute first and the smallest ones will elute last (and "middle" sized proteins will elute in the middle).
.png?revision=1&size=bestfit&width=413&height=341)
Figure 5.4.3: Gel filtration
- If your protein is unusually "small" or "large" in comparison to contaminating proteins then gel filtration may work quite well.
Where will a protein elute in a gel filtration experiment?
- There are two extremes in the separation profile of a gel filtration column.
- There is a critical molecular mass (large mass) which will be completely excluded from the gel filtration beads. All solutes in the sample which are equal to, or larger, than this critical size will behave identically: they will all eluted in the excluded volume of the column
- There is a critical molecular mass (small mass) which will be completely included within the pores of the gel filtration beads. All solutes in the sample which are equal to, or smaller, than this critical size will behave identically: they will all eluted in the included volume of the column
- Solutes between these two ranges of molecular mass will elute between the excluded and included volumes
.png?revision=1&size=bestfit&width=381&height=302)
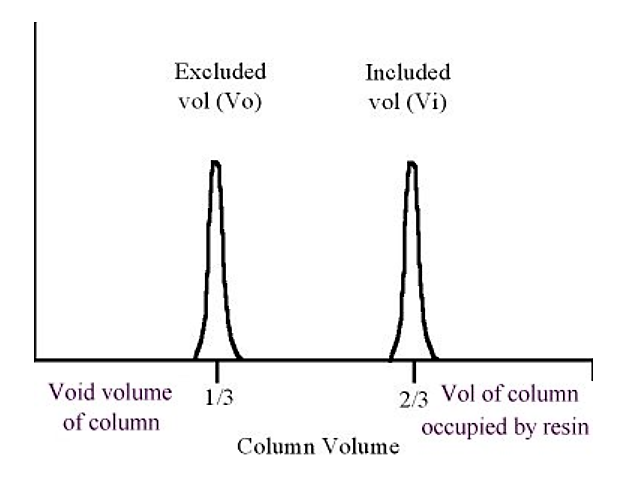
Figure 5.4.4: Protein elution in gel filtration
As a general rule of thumb, the excluded volume (Vo) is approximately equal to one third of the column volume, the included volume is approximately equal to two thirds of the column volume (the "missing" third is taken up by the volume of the resin material).
- In gel filtration the resolution is a function of column length (the longer the better)
- However, one drawback is related to the maximum sample volume which can be loaded. The larger the volume of sample loaded, the more the overlap between separated peaks. Generally speaking, the sample size one can load is limited to about 3-5% of the total column volume.
- Thus, gel filtration is best saved for the end stages of a purification ,when the sample can be readily concentrated to a small volume.
- Gel filtration can also be used to remove salts from the sample, due to its ability to separate "small" from "large" components.
- Finally, gel filtration can be among the most "gentle" purification methods due to the lack of chemical interaction with the resin.