
5.5: Gel Electrophoresis of Proteins
- Page ID
- 18151

Gel electrophoresis is used to characterize one of the most basic properties - molecular mass - of both polynucleotides and polypeptides. Here we will focus exclusively on gel electrophoresis of proteins
Gel electrophoresis can be used to determine:
- the purity of a protein sample
- heterogeneity and extent of degradation of a protein sample
- subunit composition of a protein sample
How does it work?
The underlying principle of electrophoresis is the migration property of charged species within an electric field. Thus, it is the simple behavior of opposite charges attracting. An electric field is established across the electrodes of a power supply, and charged ions move in this electric field. Note that in such an apparatus, the word "ANODE" refers to the positively charged electrode (the ANODE attracts ANIONS) and the word "CATHODE" refers to the negatively charged electrode (the CATHODE attracts CATIONS). The cathode and anode terms are consistent with their redox reaction definitions in that reduction is subsequently occurring at the cathode and oxidation is occurring at the anode:
.png?revision=1&size=bestfit&width=440&height=271)
Figure 5.5.1: Anode and cathode
Warning: read the following note only if you are curious about batteries and labeling of electrodes, otherwise, don't bother (it may only be confusing).
Note
Redox chemistry within the external power supply is driving the redox reaction at the electrodes. Note that the electrons from the anode are going to the "+" terminal of the battery. Reduction at this electrode of the battery must be occurring and is driving the oxidation at the anode of the external electrodes. If this terminal of the battery is being reduced, then it must be the cathode in the redox reaction within the battery. Thus, batteries have their cathode labeled as "+" and anode labeled as "-". Took me hours to figure this out.
Proteins are comprised of the 20 common amino acids, which include both negatively charged (i.e. acidic) side chains (e.g. aspartic acid, glutamic acid) and positively charged (i.e. basic) side chains (e.g. histidine, lysine and arginine). Thus, proteins can be charged, and will migrate in an electric field.
At this point there are a couple of things to consider:
1) Any such separation is a non-equilibrium process. By this, we mean that if we let the process continue on until some equilibrium condition is met, all the anions will be on one electrode and all the cations will be on the other. It would be better to halt the separation process at some intermediate time point to permit achieve separation:
.png?revision=1&size=bestfit&width=495&height=235)
Figure 5.5.2: Separation process
2) The other problem is that once the electric field is switched off, diffusion will cause the separated ions to move around (i.e. we will lose the separation we have tried to achieve). To solve this problem, the separation is not performed in solution, but within a matrix (i.e. a molecular mesh or network). The matrix provides a frictional component that resists diffusion. Furthermore, the friction of the matrix is an important factor in the rate of migration of the ions.
3) Note also that separation is achieved by initial application of the sample within a narrow zone (band). If the sample is initially dispersed, although the ions will move, they won't be neatly separated.
Factors that influence the rate of electrophoresis migration (Rf)
Three factors affect the rate of migration:
· Strength of the electric field, E (directly proportional to migration rate)
· Charge on the ionic species, q (directly proportional to migration rate)
· Frictional coefficient of the support matrix, f (inversely proportional to migration rate)
Rf α qE/f
These factors can be varied in the following way:
· The strength of the field is a function of the voltage of the power supply. Thus, we can vary the voltage directly. In a related issue, the voltage is proportional to the resistance across the electrodes. Current comes into play here also, but in short, it is difficult to achieve high voltage across the electrodes if the resistance is low. The resistance of a solution is inversely proportional to the ionic strength (i.e. concentration of ions). Thus, with high salt concentrations, the resistance is low, it is difficult to achieve high voltage, and the migration rate will decrease.
· The charge on an ionic molecule is a function of the pI of the molecule and the pH of the solution
If pI > pH the molecule is cationic
(migrates towards cathode)
If pI < pH the molecule is anionic
(migrates towards anode)
If pI = pH the molecule is neutral
(no migration in electric field)
We can therefore alter the migration rate (and possibly the direction of migration) by altering the pH of the solution
· The frictional coefficient of the matrix can potentially be altered. The matrix is typically a polymer network (see below), referred to as a gel, and the frictional coefficient can be increased by increasing the polymer concentration
The gel matrix for gel electrophoresis of proteins
Gel electrophoresis of proteins almost exclusively utilizes polyacrylamide. This is a polymer comprised of two covalently-linked components:
- acrylamide
- bis acrylamide.
The bis acrylamide is essentially a cross-linking component of the acrylamide polymer. A typical value for the acrylamide:bisratio is 19:1 and the total acrylamide concentration in the gel affects the migration of proteins through the matrix (i.e. determines the frictional coefficient).
- High molecular mass proteins are separated using low frictional coefficient (i.e. low concentrations) of polyacrylamide.
- Low molecular mass proteins are separated using high frictional coefficient (i.e. high concentrations) of polyacrylamide.
- Although we now have everything in place to perform a gel electrophoresis experiment to separate proteins, there is another consideration that often is undesireable (although sometimes useful). This situation is the fact that different proteins have different charges for a given value of pH. Thus, proteins will migrate as a function of both their mass (big ones move slowly) and their overall net charge.
- If there were some way to cause each protein to have an identical charge to mass ratio, we could separate a mixture of proteins based only upon mass effects
The role of sodium dodecyl sulfate detergent in polyacrylamide gel electrophoresis
Sodium dodecyl sulfate (SDS; also known as "laurel sulfate") is an ionic detergent with the following structure:
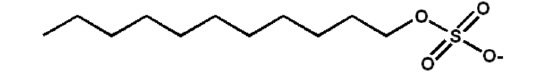
Figure 5.5.3: Sodium laurel sulfate structure
- The SDS binds, via hydrophobic interactions, to the proteins in a stoichiometry approximately proportional to the size of the protein (i.e. a small protein will bind a few molecules, and a large protein will bind a lot of molecules of SDS)
- Due to the charged nature of the SDS molecule the proteins thus will have an approximate constant charge to mass ratiodue to the charge provided by the SDS, and will migrate through the gel at a rate proportional to their molecular mass
- The proteins migrate towards the anode since the charge on the SDS is negative under all pH conditions except highly acidic.
Protein gels are usually performed under denaturing conditions, meaning that the sample preparation involves heating the protein in the presence of SDS to fully unfold the protein and permit binding of SDS throughout the length of the polypeptide. Once SDS has been bound, the characteristic pI values of the proteins is no longer relevant; the protein takes on a negative charge, and each protein has essentially the same charge to mass ratio.
Migration rate, protein mass, and the % acrylamide in the gel
The greater the percent acrylamide in the gel support, the greater the frictional coefficient, and the slower the migration rate. If the proteins to be separated are of a high molecular mass, and if the % gel is high, the proteins may not even enter the gel (due to overwhelming friction). Thus, it is essential to match the % gel to the mass of the proteins being separated. The following table provides a general guideline:
|
Acrylamide |
Range of separation of Polypeptides (length in amino acids) |
|
8% |
25-200 kDa |
|
10% |
15-100 kDa |
|
12.5% |
10-70 kDa |
|
15% |
6-60 kDa |
|
20% |
4-40 kDa |
In setting up the SDS PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) experiment we need to know when to stop the experiment (since it is not an equilibrium process). This is somewhat difficult to determine since proteins (even with SDS bound) do not absorb in the visible spectrum (i.e. we cannot simply look at the gel to determine when the proteins have been separated). Therefore, it is common to include in the protein sample a small anionic dye molecule (e.g. bromophenol blue):
- The size of the dye molecule is chosen to be very small so that there is essentially no friction coefficient with the gel
- The dye is chosen to be anionic so that it will migrate in the same direction as the protein/SDS complexes (i.e. towards the anode)
- Since the dye is anionic, and small, it will migrate the fastest of any component in the mixture to be separated
- The dye molecule is also chosen so as to absorb in the visible spectrum (and, hence, be visually detectable while the gel is running)
- The protein/SDS/dye mixture is loaded on the top of the gel (i.e. cathode side) and when the dye molecule (the "dye front") reaches the bottom of the gel, the power is turned off and the experiment halted
Visualization of the separated proteins
Although the gel support provides some friction to molecular motions, as soon as the power is turned off the separated protein bands will begin to diffuse (they are freely soluble in aqueous solution). To prevent this, the gel is treated with an acetic acid and methanol solution which causes almost all proteins to precipitate (become insoluble). This is called "fixing" the gel. Now the separated proteins will not diffuse.
The fixed proteins are, however, still invisible and must be visualized by staining. A common protein stain is Coomassie Brilliant Blue R-250 (related to the dye used previously in the Bradford assay). The fixed gel is incubated in a solution of "Coomassiestain" and then the stain is washed out of the gel by incubation in a weak solution of acetic acid and methanol. The stain will not bind to the acrylamide, and will wash out (leaving a clear gel). However, it remains strongly bound to the proteins in the gel, and these take on a deep blue color.
Determination of molecular mass
With SDS treatment, the proteins will migrate as a function of their molecular mass. The approximate molecular mass of the separated proteins is therefore a function of their migration distance. If a series of proteins with different and known molecular masses is analyzed on an identical gel, under identical conditions, then a standard curve can be established that can be used to determine the molecular mass of an unknown protein. A typical "molecular weight standard" includes the following mixture of proteins:
|
Protein |
Molecular Mass (kDa) |
|
Phosphorylase B |
94 |
|
Bovine Serum Albumin |
67 |
|
Ovalbumin |
43 |
|
Carbonic Anhydrase |
30 |
|
Soybean Trypsin Inhibitor |
20.1 |
|
a-Lactalbumin |
14.4 |
The molecular mass is quantified as follows:
- Measure (in cm) the migration distance of the dye front
- Measure (in cm) the migration distance of the protein (will always be less than the dye front)
- Divide the protein migration distance by the dye front distant to get the relative mobility value (always < 1)
- Plot the relative mobility value (along x-axis) versus the log of the molecular mass (along y-axis)
- The relationship between the relative mobility and the log of the molecular mass should be a linear function - thus, providing a standard curve against which the molecular mass of an unknown protein can be determined from its relative mobility
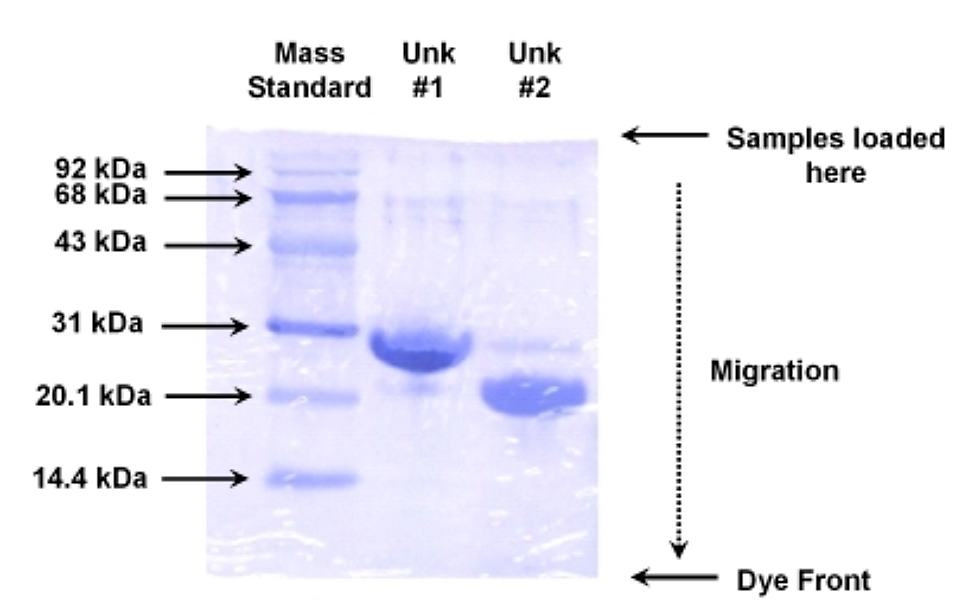
Here is an example of a molecular mass standard, and two unknown samples, analyzed on a 10% SDS PAGE stained with Coomassie:
.png?revision=1&size=bestfit&width=457&height=289)
Figure 5.5.4: Example gel electrophoresis
The calculation of migration distances, relative mobility and relationship to Log of the molecular mass yields the following:
.png?revision=1&size=bestfit&width=463&height=383)
Figure 5.5.5: Relative mobility and molecular mass
- Unknown #1 has a value of 1.43 for the log of the molecular mass, or a mass of ~26.9 kDa
- Unknown #2 has a value of 1.33 for the log of the molecular mass, or a mass of ~21.4 kDa
- A visual check of the unknowns versus the standards in the above gel indicate the calculations appear to be correct
There are other variations of SDS PAGE including:
- Discontinuous gels (i.e. a composite of one gel on top of another) to achieve "focusing" of protein bands (i.e. sharper bands, yielding greater sensitivity and resolution)
- Gradient gels (instead of pouring a single % polyacrylamide gel, pour a gradient with a high concentration at the bottom, and a lower concentration towards the top. This permits resolution of a wider range of molecular mass samples)
- Silver staining (a more sensitive type of stain in comparison to Coomassie - allowing detection of lower concentrations of proteins)
- Transfer of resolved protein bands to a secondary support (e.g. nitrocellulose) for probing with other reagents (i.e. antibodies)
- PAGE (no SDS) of native proteins (to allow detection of non-covalent complexes of two or more proteins - since SDS will disrupt such complexes)
- SDS PAGE with the addition of reducing agents (e.g. b-mercaptoethanol, dithiothreitol, etc.). These reagents will reduce disulfide bonds and separate polypeptide chains that are connected by such bonds
However, the above information covers the basics.