
11.5: Active Transport
- Page ID
- 51045


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In the previous sections, we explored facilitated diffusion, diffusion through channels, and diffusion through larger pores. In each case, once a carrier/permease protein was available, or a channel (gated by ligand binding, change in membrane potential, lipid binding, or mechanical forces) or a pore formed, solute flows down a chemical gradient (facilitated diffusion) or electrochemical gradient in a thermodynamically favored process. But what about moving solutes from low to high concentration, against a concentration gradient, which would be necessary to capture the last bit of a vital nutrient or energy source? Active transport does just that, but it requires an energy source to do so. Many different types of chemical species are actively transported across the cell and organelle membranes, including sugars, amino acids, (deoxy)nucleotides, metabolites (like carboxylic acids),
Type of Active Transport
For active transport to occur, a membrane receptor is required which recognizes the ligand to be transported. Of major interest to us, however, is the energy source used to drive transport against a concentration gradient. The biological world has adapted to use almost any source of energy available.
ATP hydrolysis: One would expect that this ubiquitous carrier of free energy would be used to drive active transport. This is one of the predominant roles of ATP in the biological world. 70% of all ATP turnover in the brain is used for the creation and maintenance of a Na and K ion gradient across nerve cell membranes using the membrane protein Na+/K+ ATPase.
Energy released by oxidation: You may have encountered this in previous biology courses. Active transport of protons driven by oxidative processes is exergonic. In electron transport in respiring mitochondria, NADH is oxidized as it passes electrons to a series of mobile electron carriers (ubiquinone, cytochrome C, and eventually dioxygen) using protein complexes in the inner membrane of the mitochondria. The energy lost in this thermodynamically favored process is coupled to conformational changes in the complex which caused protons to be ejected from the matrix into the inner membrane space. One can imagine a series of conformation-sensitive pKa changes in various side chains in the complexes which lead in concert to the vectorial discharge of protons.
Light: Photosynthetic bacteria have a membrane protein called bacteriorhodopsin which contains retinal, a conjugated polyene derived from beta-carotene. It is analogous to the visual pigment protein rhodopsin in retinal cells. Absorption of light by retinal induces conformation changes in the retinal and bacteriorhodopsin, which leads to the vectorial discharge of protons.
The collapse of an ion gradient: The favorable collapse of an ion gradient can be used to drive the transport of a different species against a concentration gradient. We have already observed that the collapse of a proton gradient across the inner mitochondria membrane (through F0F1ATPase) can drive the thermodynamically unfavored synthesis of ATP. The collapse of a proton gradient provides a proton-motive force that can drive the active transport of sugars. Likewise, a sodium-motive force can drive the active transport of metal ions. Since the energy to make the initial ion gradients usually comes from ATP hydrolysis, ATP indirectly powers the transport of the other species against a gradient.
Active transport can be divided into two classes:
- primary - driven by "primary" energy sources, which for us means ATP or photons
- secondary - driven by coupled transport (also called cotransport) of solutes down a concentration gradient, which provides the thermodynamic force to move a solute "uphill".
Active transporters can also be divided into classes based on the direction of movement of the solute and any cotransported solute into uniport (no cotransported solute), symport (solute and cosolute transported in the same direction) and antiport (solute and cosolute transported in the opposite direction) as shown in Figure \(\PageIndex{1}\).

Examples of these different mechanisms can be found in facilitated diffusion as well as active transport. Examples of these types include:
- uniport: An often-used example is GLUT 1, which catalyzes the FACILITATED DIFFUSION (not active transport) of glucose down a concentration gradient. A somewhat complicated example of a uniport active transporter is the Na+/K+ ATPase, which moves Na+ ions out of a cell and K+ into a cell, both against concentration gradients. The movement of one ion does not provide the chemical potential thermodynamic driving force to move the other. Rather the hydrolysis of ATP is required. So this protein can be called a uniport active transporter for two different ions.
- symport: Another glucose transporter, the glucose symporter (SGLT1), found in the small intestines, heart, and brain, cotransports one glucose or galactose for every two Na+ ions that move into the cells down a concentration gradient.
- antiport: The sodium-calcium exchanger pumps out one Ca2+ ion (low to high concentration) driven by the influx of three Na+ into the cell. This keeps Ca2+ low in the cytoplasm.
If the species moved is charged, two other terms are used:
- electrogenic - a net electrical imbalance is generated across the membrane by symport or antiport of charged species
- electroneutral - no net electrical imbalance is generated across the membrane by symport or antiport of charged species
In this chapter section, we will explore examples of several types of active transporters. All are polytopic with alpha-helical transmembrane domains, which through a series of conformational transitions can move chemical species that are bound to the receptor or channel protein to move through the membrane through the creation of transient openings in the protein. We will end with another look at the nuclear pore complex and see how it allows the movement of large proteins and RNA molecules through its pore through a very different type of mechanism. In that case, it is not active transport since the relative concentration of the transported species on either side of the pore is less important than the mechanism by which certain species are targeted to move into and out of the nucleus.
Major Facilitator Superfamily (MFS) Transporters
There are about 74 different families of active transporter in the Major Facilitator Superfamily (MFS), which is the largest of secondary transporters using symport or antiport mechanisms. There are also uniporters within this family. Different members of the MFS move a diverse array of solutes, including sugars, nucleotides, peptides, and drugs across membranes against a concentration gradient. All have two membrane domains consisting of six transmembrane helix bundles that pack to give an apparent two two-fold rotational axis of symmetry (also called pseudo C2 axis). The ligand to be transported binds between the two domains in a central cavity potentially open to either side of the membrane. Figure \(\PageIndex{2}\) panel A (top) below shows a cartoon of the 12 helices and 6-helix bundles. Helix 1 and 4 in domain one and the analogous ones in domains 7 and 10 are involved in ligand movement through the membrane. Correspondingly, helices 2, 5, 8, and 11 are involved in domain-domain interactions. The bottom figure (B) shows two views of the protein with the ligand in spacefill rendering.

As structure mediates function, they all display a similar "clamp and switch" mechanism, in which solute binds to the protein in an open (to binding) conformation, leading to conformation changes forming a closed form, followed by further conformational changes which cause the complex to open to the other side of the membrane. Figure \(\PageIndex{3}\) shows hands opening, closing, and reopening in the other direction as an analogy for this "clamp-switch" mechanism.

We will describe two different transporters in this family.
Human monocarboxylate transporter (7BP3)
These proton-linked monocarboxylate transporters (MCTs) move many monocarboxylates (including lactate, pyruvate, and the ketone bodies acetoacetate, beta-hydroxybutyrate and acetate) out of the cell, driven by the flow of protons across the membrane in the same directions (symport). In cells engaging in glycolysis, a fundamental energy utilization pathway, it catalyzes the high-affinity transport of pyruvate, the end product of glycolysis, and lactate, a reduced derivative of pyruvate, out of the cell. In cells that are not engaged in glycolysis, their activity is effectively turned off. The flux of pyruvate/lactate has steep dependence on their concentrations, allowing the protein to act as an on-offf switch for transport. A step dependency of activity on concentration is a hallmark of cooperative interactions. Structural analyzes show conformational changes in the interface of the dimeric form of the transporter.
We will see when we study glycolysis, the conversion of the oxidized pyruvate to its reduced form lactate requires a reducing agent, in this case, NADH, which is oxidized back to NAD+, an oxidizing agent required for glycolysis to continue. When pyruvate is high, NADH is also high, allowing for the conversion of pyruvate to lactate, which can be exported out of the cell. The interconversion is described by this reaction:
Pyruvate + NADH ↔ Lactate + NAD+
Using MCT 2 to remove lactate from the cell pulls the above reaction to the right, regenerating NAD+, and allowing glycolysis to continue.
Figure \(\PageIndex{4}\) shows an interactive iCn3D model of the inward open human monocarboxylate transporter 2 (7BP3).
.png?revision=1&size=bestfit&width=430&height=303)
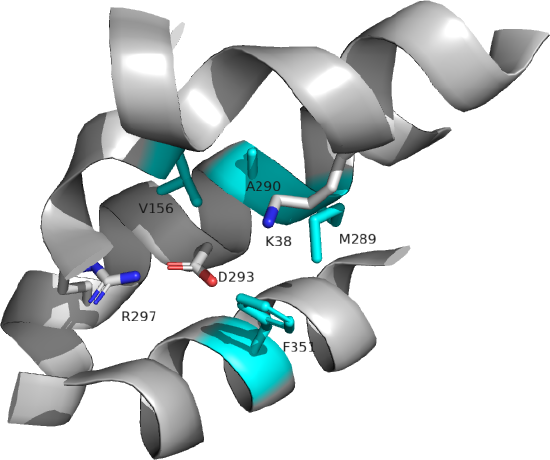
Several key residues are shown. Aspartic acid 293 (side chains in red sticks) is conserved in all MCTs studied and is likely involved in H+ transport. It is surrounded by Val 156, Met 289, Ala 290, and Phe 351 (side chains in cyan sticks), all nonpolar amino acids which would elevate the pKa of Asp 293, allowing it to stay protonated until a conformational change would alter its environment, leading to proton release. Two positive charged residues, Lys 38 and Arg 297 (side chains in blue sticks) are involved in substrate interactions. Lys 38 faces to the outside of the cell in the inward open conformation. Figure \(\PageIndex{5\) below shows the same amino acids in a zoomed view. The amino acids are labeled.
Lactose Permease (Lactose-proton symport)
This E. Coli protein is also a symporter, which uses the movement of H+ down a concentration gradient to drive lactose into E. Coli to capture as much lactose - an energy source - as possible. Deprotonation of a protonated Glu 269 leads to ligand binding as the protein has no binding site in the protonated site. Lactose induces the formation of its binding site. Figure \(\PageIndex{6\) below shows the change in conformation going from a more acidic pH (5.6, PDB 2CFP) to a more neutral pH (6.5, PDB 2CFQ). One lactose is transported from one H+, which moves down a concentration gradient. Lactose moves from the outside periplasm to the cytoplasm.
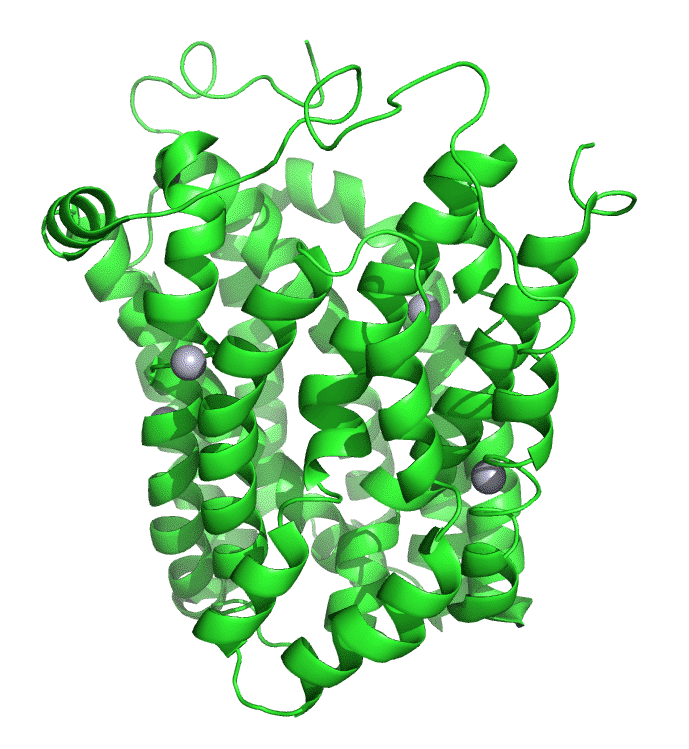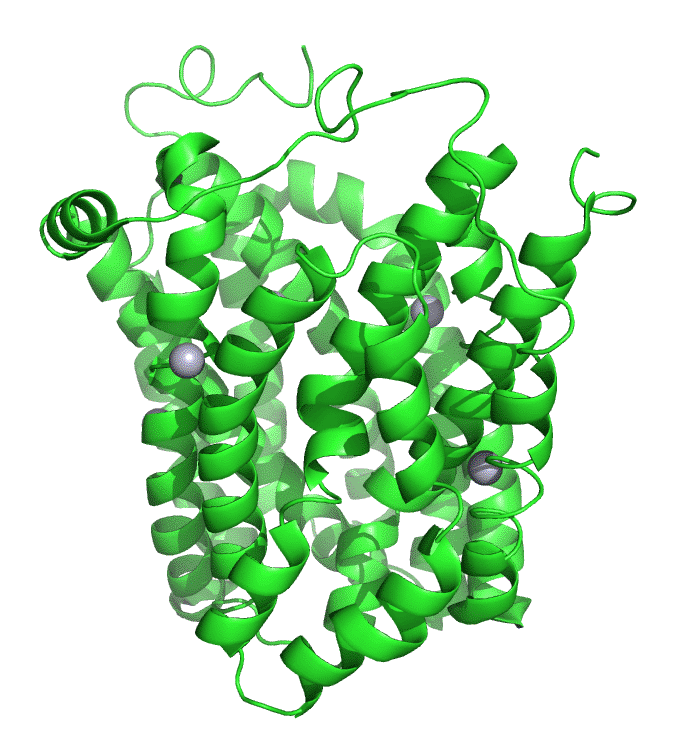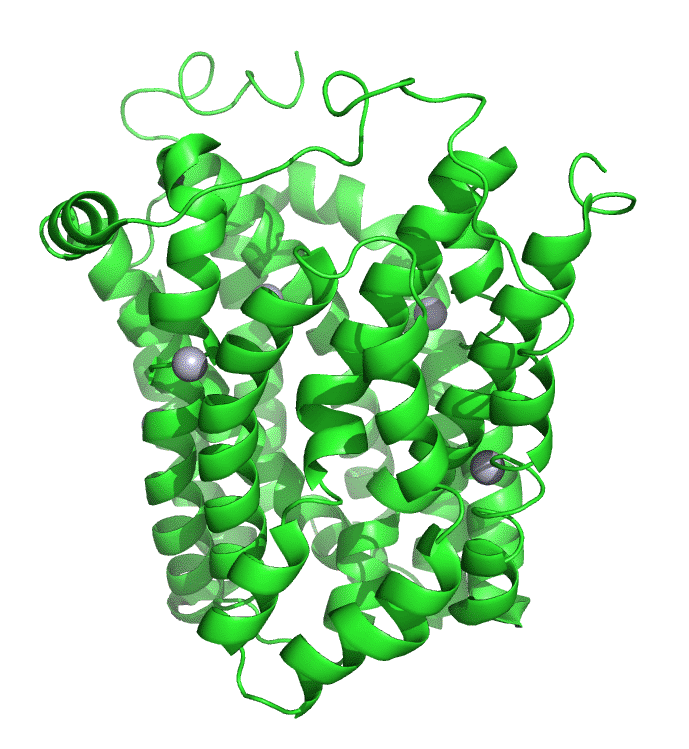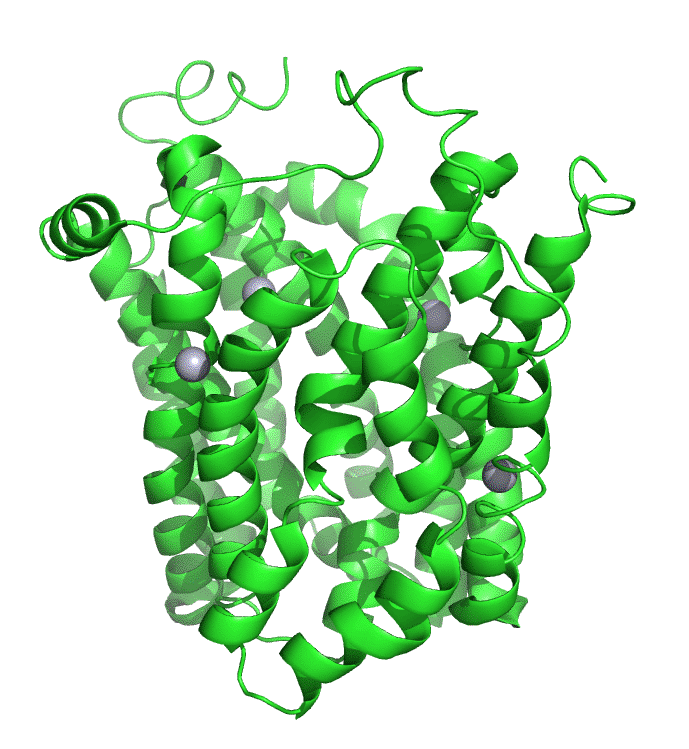
Figure \(\PageIndex{7}\) shows an interactive iCn3D model of lac permease (1pv7), which highlights three residues essential for lactose binding (Glu 126, Arg 144, and Glu 269 in stick) and three involved in proton transfer (Arg 302, His 322 and Glu 325 in spacefill).
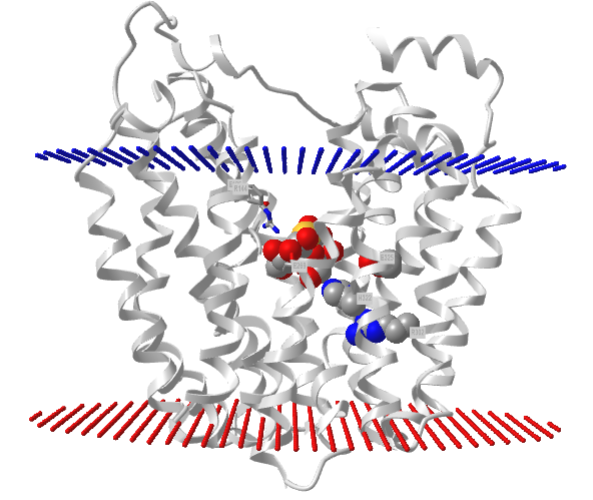.png?revision=1&size=bestfit&width=412&height=347)
It has a bound nonhydrolyzable lactose analog (beta-D-galactopyranose-(1-1)-1-thio-beta-D-galactopyranose), shown in spacefill, in the center hydrophilic cavity. The two 6-helix bundles, which give pseudo 2-fold symmetry are evident. The protein is in the open-to-inward conformation. (The blue dummy atoms represent the inner leaflet.)
Figure \(\PageIndex{8}\) is a video of a molecular dynamics simulation showing lactose moving through lactose permease.
Figure \(\PageIndex{8}\): Molecular dynamics simulation showing lactose moving through lactose permease. https://www.ks.uiuc.edu/Gallery/Movi...annelProteins/
As with other members of the MFS transporters, lactose permease has 12 transmembrane helices that form two bundles. The lactose binding site is between the two bundles. When the protein is in the open-to-out conformation, it can bind protons, most likely at an exposed His. This enables lactose binding, which is followed by a conformational change to the open-to-in form. The protonated His loses a proton and lactose dissociates into the cytoplasm. Movement of both H+ and lactose together must occur for transport.
Structural work by Singh et al on the Leu Transporter (LeuT), a member of the solute carrier 6 or sodium-coupled transporters, which is an active transporter requiring movement of Na ion into the cells to power the uptake of Leu, shows an "open-to-out" and occluded binding state for ligand (Leu). Tryptophan, a competitive nontransportable inhibitor binds to the open-to-out state but is too large for the obligate occluded state so it is not transported. Figure \(\PageIndex{9}\) shows inward open, closed, and outward conformations of different major facilitator superfamily transporters.

Another member of the MFS Transporter family is LeuT, the leucine transport, which is similar to the neurotransmitter sodium symporter. Figure \(\PageIndex{10}\) shows the transition between two conformational states. The first is the outward open leucine transporter (LeuT). This initial state has bound tryptophan, a competitive inhibitor, which is too big to transport, which traps the bound state in the open conformation. The second state has bound Leu and is closed. The conformational changes are subtle but sufficient to allow transient binding, occlusion, and expulsion to the other side of the membrane (not shown). Note also the two bound Na+ ions (purple) clearly show that active transport through this transporter is driven by Na+ ions. The ions are farther apart in the open state allowing access to the amino acid for transport.

Membrane ATPases
Many active transporters power the uphill transport of solute through coupled exergonic cleavage of ATP. The largest family of is the P-type ATPase, which is used to transport a host of different solutes, including ions (Cu, Zn, Mn, Mg, Ca, Na, K, Cd, Co, Pb, Ni ,and H) and phospholipids (from one leaflet to another by translocases like flippases). They convert chemical energy (from cleavage of phosphoanhydride bonds) to electrochemical potential energy. Over 160,000 P-type ATPases from prokaryotes and eukaryotes are known so they are highly prevalent and evolved early in time. They should be understood at both a structural and thermodynamic level.
How is energetic coupling achieved? An early model proposed by Albers and Post suggested that energetic coupling occurred by the covalent transfer of a terminal phosphate (Pi) from ATP to an Asp in the protein channel to form a covalent Asp-Pi intermediate, which is an example of a mixed anhydride. This is illustrated in Figure \(\PageIndex{11}\).

This reaction is disfavored thermodynamically since hydrolysis of ATP to form ADP proceeds with a less negative ΔG0 than the same reaction with a mixed anhydride. However, ultimately hydrolysis of the mixed anhydride in the next step would make the overall coupled reactions favorable.
Another feature of the Post-Albers model is that the phosphorylated channel, P-ATPase, has two conformations:
- a high-affinity cation inward (cytoplasm) facing cation binding site called E1
- a low-affinity cation outward (extracellular or luminal) facing cation binding site called E2
Figure \(\PageIndex{12}\)s a more detailed (A) and simpler (B) set of chemical equations to represent the different states of E1 and E2 in the Post-Albers model.
The next three figures are from the following reference: Zhang, X.C., Zhang, H. P-type ATPases use a domain-association mechanism to couple ATP hydrolysis to conformational change. Biophys Rep 5, 167–175 (2019). https://doi.org/10.1007/s41048-019-0087-1. Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/

- The top line in both shows the reactions of E1 (high affinity) with the substrate (ions) and ATP. Since E1 is phosphorylated by ATP, it is considered a kinase.
- The bottom line in both shows the reactions of E2 (low affinity) with the substrate (ions). Since E2 is dephosphorylated, it is considered a phosphatase.
S1 and S2 differ for specific P-type ATPase.
Detailed Structures
Now let's look at the structures of a specific P-type ATPase, the sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (or more simply a Ca2+ pump), which catalyzes the active uphill transport of Ca2+ from the cytoplasm back into the lumen of the sarcoplasmic reticulum (SR) in muscle cells and acts in the regulation of striated muscle contraction. The actual free concentration of Ca2+ in the SR is about 390 μM compared to 0.1-0.2 μM in the cytoplasm. On muscle excitation, Ca2+ floods out into the cytoplasm but must be actively transported back into the SR by the Ca2+ pump.
Here is the simplified chemical reaction: ATP + Ca2+(cyto) + H2O → ADP + Ca2+(s. rect) + H+ + Pi.
We'll stick with a cartoon representation of each of the various structure states of this calcium pump, as illustrated in Figure \(\PageIndex{13}\) (also from Zhang et al).

Again, different states of E1, the kinase state, are shown in the top line, while E2, the phosphatase state, is shown in the bottom line. PDB IDs are shown for each state. Ca2+ is shown as a cyan sphere. Note how the cytoplasmic N (nucleotide-binding), P (phosphorylation), and A (actuator) domains and the single transmembrane domain, colored to show the C-terminal (CTM) and N-terminal (NTM). are represented in the cartoon version to the right. P represents the phosphorylated Asp 351 mixed anhydride. The asterisk * denotes a transition state for the phosphorylation of Asp 351 (top line) and its dephosphorylation (bottom line).
Ca2+ moves from the cytoplasm to the lumen of the SR. Note that the first E1 state is open inward (to the cytoplasm) while the first E2 state is open outward (to the lumen of the Sr, where the bulk of the Ca2+ ions are stored. This mimics the open-in and open-out states of the Major Facilitator Superfamily (MFS) channels discussed above.
Detailed Thermodynamics
Zhang et al provide two different views of the thermodynamics of Ca2+ ion pumping into the SR as shown in A (domain dissociation) and B (classic) in Figure \(\PageIndex{14}\). A quick view of the two "energy landscapes" show:
- Both go from higher to lower free energy as expected since ATP hydrolysis powers the process.
- They differ mostly in the energetic coupling steps

In A, the actual energy released in ATP hydrolysis occurs when the Asp 351-Pi mixed anhydride is dephosphorylated (ΔGdeph), which is coupled to the dissociation of the P and A (* actuator) domains. This is shown in the structural cartoon near the last step in the complete cycle.
In the cartoon diagram, 6 states are represented. Any mechanism can be broken down into more and more states on a more complete description of the actual mechanism. For example, ATP cleavage to ATP can be represented by three states, ATP, ADP, and Pi, or many more if intermediates and transition states are included, the complete Ca2+ transport cycle in the thermodynamics diagram is broken down into 12 states, represented by 8 horizontal solid lines _____ and 4 dashed ------ lines. Vertical lines with arrows show transitions between states.
- Green arrows → show changes in free energy due to the relative stability (chemical potential) of ATP.
- Red arrows → show changes in free energy due to the relative stability (chemical potential) of the substrate ions
- Cyan arrows → show changes in free energy due to the electrical potential of the substrate ions
ΔGL are for steps involving loading of reactants, while ΔGR are for release. In both diagrams, the conversion of state E1.S.ATP to E1P.S is uphill as we predicted for the formation of the
On the far right of each graph are identical sets of large unfilled arrows (since ΔG is a state function) as the beginning and ending free energies of free E1 must be pathway (i.e. mechanism) independent. The two sets of upward unfilled arrows (red for the change in the chemical potential of the substrate ions and cyan for the change in the electric potential of the substrates are both positive as the ions move uphill from a lower to high electrochemical potential. The unfilled downward green arrow shows the change in free energy (or chemical potential) for ATP hydrolysis. Since energy cannot be created or destroyed, the narrow black arrow (Qx) represents energy that is dissipated in the reaction cycle. The starting and ending states are identical (i.e., E1), only being differed by the energy dissipation (denoted as QX) of the P-ATPase transporter during one functional cycle.
The are other types of membrane ATPase whose structure and function differ from the P-type described above. Some run in reverse to synthesize ATP. They also vary in substrate ions. Here are several different types.
- F-ATPases (ATP synthases, F1F0-ATPases): These are used to synthesize ATP and are powered by the collapse of a H+ gradient. They are found n mitochondria, chloroplasts, and bacterial cell membranes. We will discuss these more in chapters on mitochondrial oxidative phosphorylation and photosynthesis in chloroplasts.
- V-ATPases (V1V0-ATPases): These are used to pump protons into organelles to acidify them (ex. lysosome) and are also found in bacteria.
- A-ATPases (A1A0-ATPases): These are used to synthesize ATP in Archaea.
- E-ATPases: These are found on the cell surface and hydrolyze extracellular nucleotide triphosphate.
Let's now look at some special features of one P-type ATPase that transports both Na+ and K+ ions and is important in establishing their intracellular and extracellular concentration in neurons, so they are critical for neuron function.
Na+/K+ ATPase
This protein keeps the K+in and Na+out high compared to their respective concentrations on the other side of the neural cell membranes. ATP and 3 Na+ ions bind to the cytoplasmic domain of the enzyme in the E1 conformation. As described more generally above, in the presence of Na ions, the bound ATP is cleaved in a nucleophilic attack by an Asp side chain of the protein. Hence, the protein is a Na+-activated ATPase or kinase. The phosphorylated enzyme changes conformation to the E2 form in which Na+ ions are now on the outside of the cell membrane, from which they dissociate. The phosphorylated protein in conformation E2 now binds 2 K+ ions on the outside, which activates hydrolysis of the Asp-PO3 mixed anhydride link. The dephosphorylated protein is more stable in the E1 conformation, to which it changes as it brings K+ ions into the cell. Hence this protein is an electrogenic antiporter. P-Type transporters are inhibited by vanadate (VO43-), a transition state analog of phosphate. Transport mediated by P-type membrane proteins can, in the lab, be used to drive ATP synthesis.
Detailed kinetic analysis of ATP and VO43- interactions show there are low-affinity and high-affinity sites for each on Na/K ATPase. The high-affinity vanadate site appears to be the same as the low-affinity ATP site, which suggests that vanadate binds tightly to the E2 form of the enzyme. The low-affinity vanadate site appears to be the same (based on competition assays) as the ATP site, which is probably the E1 form. Hence vanadate binds preferentially to the E2 form which would inhibit the transition to the E1 form. Vanadate also inhibits phosphatases, enzymes that cleave phosphorylated Ser, Thr, and Tyr phosphoesters in proteins. This supports the notion that vanadate binds preferentially to the E2 form, which has a phosphoanhydride link (Asp-O-phosphate) that is hydrolyzed, promoting the conversion of E2 back to E1. Vanadate is probably a transition state analog inhibitor in that it can readily adopt a trigonal bipyramidal structure, mimicking the transition state for cleavage of the tetrahedral anhydride bonds of ATP and Asp-O-PO3.
Figure \(\PageIndex{15}\) is a YouTube animation of the Na+/K+ ATPase from xx. Permission Question?
Figure \(\PageIndex{15}\): Animation of the Na+/K+ ATPase pump
These interactions are depicted in Figure \(\PageIndex{16}\)below (after Stryer 4th ed)

ABC Transporters
The proteins comprise one of the largest families of membrane proteins with seven main families (ABCA to ABCG). Up to 3% of all bacterial proteins encode proteins associated with the ABC transporters. Different ABC transporters move a variety of required chemical species, from small (ions, sugars, amino acids, nucleosides, vitamins) to large (peptides, lipids, oligonucleotides, and polysaccharides) into the cell. They also remove toxic (to the cell) molecules such as xenobiotics (molecules foreign to the cell like drugs, toxins, etc) and potentially toxic metabolites. All of these require ATP hydrolysis. Moving toxic molecules out of the cell is beneficial to the health of the cell, but in the case of a tumor cell, not to the benefit of the organism. TAs in other active transporters using ATP, its hydrolysis leads to two different conformations, open-outward and open-inward. All ABC transporters have a LSGGQ amino acid sequence in the NBD. They also have a phosphate-binding loop (P-loop or Walker A motif). Most eukaryotic ABC transporters move solutes from the inside to the outside of the cell.
The various structures within the ABC transporter superfamily are shown in Figure \(\PageIndex{17}\). The transmembrane domain (TMD, in green) and the nucleotide-binding domain (NBD, blue) are present in most versions of the gene. PK represents prokaryotic and EK eukaryotic organisms (shown at the very right for each).

The ABC transporter genes denoted as full structures have 2 TMDs and 2 NMDs while half structures have one of each. Some of the genes encode either a single NBD or a single TMD (prevalent in prokaryotes, along with half structures). The ABC2 structure has only two NBDs. The single structure represents the ABC transporter gene with a single TMD or NBD; ABC2 structure represents the ABC transporter gene with only two NBDs. The families possessing certain structures are listed on the right. TMDs typically have 6-10 transmembrane α-helices. Those that are involved in the export of chemical species have six.
Structural cartoons representing domain and ligand binding for a variety of ABC transporters are shown in Figure \(\PageIndex{18}\) below.

(A) shows an inward-open drug (D) exporter. Binding of 2 ATPs cause dimer formation between the two NBD domains, resulting in a conformation change to outward facing which allows dissociation of the drug. Hydrolysis of ATP enables the release of ADP/Pi and dissociation of the 2 NBD domain, which results in conformational change to the inward-open form.
(B) shows the delivery of a substrate (S) from its binding protein (ex. part of the ABC transporter complex MalEFGK involved in maltose/maltodextrin import) in the periplasm of E. Coli for delivery into the cell.
(C) shows an outward-open variant of (B) (ex. part of the Vitamin B12 import system permease protein BtuC)
The mammalian protein multi-drug resistance (MDR), also known as P-glycoprotein, Phospholipid transporter ABCB1 or ATP-dependent translocase ABCB1, is an example of an ABC Transporter. It moves both drugs across the membrane as well as phospholipids, including phosphatidylcholine, phosphatidylethanolamine, ceramides, and sphingomyelins, from the inner to the outer leaflet of the membrane. The gene for this protein is often mutated in tumor cells which moves chemotherapeutic drugs from the cell.
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of the structure of the mouse P-glycoprotein (4M2T) bound to a cyclic peptide inhibitor in the inward open conformation is shown below. Most proteins bind substrates specifically but P-glycoprotein binds them quite indiscriminately which makes this protein so useful in pumping drugs out of the cell.
_bound_to_an_cyclic_peptide_inhibitor_in_the_inward_open_conformation.png?revision=1)
The iCn3D model below shows the conserved (mouse and human) transmembrane domain aromatic amino acids (H60, F71, T114, F299, Y303, Y306, F332, F339, F724, F728, F766, F938, Y949, F953, F974, F979) involved in the transport pathway in colored sticks. The inhibitor (cyclic-tris-(S)-valineselenazole; QZ59-SSS) is shown in spaceiflll. The consensus LSGGQ (527-531 and 1172-1176) sequences in the nucleotide-binding domain are shown in colored spheres.
Here is a view showing just the transmembrane domain with the conserved aromatic amino acids again.
Figure \(\PageIndex{20}\) shows an interactive iCn3D model of just the transmembrane domain with the conserved aromatic amino acids of the mouse P-glycoprotein bound to a cyclic peptide inhibitor in the inward open conformation (4M2T).
..png?revision=1&size=bestfit&width=370)
Another look at the Nuclear Pore Complex
We examined the incredibly complicated structure of the nuclear pore in the previous section. Both small molecules and large proteins (synthesized in the cytoplasm) and RNAs (synthesized in the nucleus) move through its large pore. The movement of solutes through its gaping pore does not require either ATP cleavage in a primary active transport or the collapse of a chemical gradient in a secondary active transport process. Then why study it in this chapter section? It turns out that the regulation of the process requires GTP cleavage and a gradient of a particular protein called Ran.
What's fascinating about the nuclear pore is its selectivity for protein transfer across the pore. What proteins are allowed in and out? What is the origin of the specificity? The specificity is determined in part by a protein family called the karyopherin-βs (22) of receptors, which bind and transport nuclear proteins. There are two types of these nuclear transport receptors or transport factors:
- Importins facilitate the movement of proteins into the nucleus (ex. nuclear proteins like histones, DNA and RNA polymerases, etc).
- Exportins facilitate the movement of RNA (except mRNA) out of the nucleus.
Large proteins destined to be moved through the nuclear pore are called cargo proteins. They have a molecular signal that differentiates them from molecules that should stay in the cytoplasm or move into organelles or be secreted from the cell. The signals are called:
- NLS or nuclear localization sequences
- NES or nuclear export sequences
There is no obvious NLS consequence and different motifs are used by different cargos. A classical NLS is enriched in Lys and Arg residue. Others appear enriched in Pro-Tyr or Ile-Lys. Large domain structures might also participate in the NLSs. Prediction programs are used to identify clusters of lysines and arginines with gaps between the clusters. Classical NESs appear to be rich in leucines in an 8-15 amino acid sequence with regularly spaced conserved hydrophobic amino acids.
A third player, RAN (Ras-related nuclear protein), is involved that determines which way the transport receptor:cargo complex moves. Ran is a small protein that binds and can hydrolyze GTP to GDP. It is a member of the small G proteins that are GTPase.
Ran itself can run (a pun) or move across the nuclear membrane and is found in both the cytoplasm and nucleus. What determines which way a Cargo:receptor complex goes? It depends on whether GTP or GDP is bound to RAN! There are higher concentrations of RanGTP in the nucleus and lower concentrations in the cytoplasm.
- Importins bind cargo proteins with a NLS in the cytoplasm where RanGTP is low, move into the nucleus, and release the cargo protein when the abundant RanGTP displaces the bound cargo protein
- Exportins bind both a cargo protein with a NES and Ran:GTP to form a ternary Exportin:Cargo:RanGTP complex in the nucleus. This moves into the cytoplasm, where the Ran bound GTP is hydrolyzed to GDP, causing the complex to dissociate, freeing the exported cargo protein from the complex.
Importins are dimeric structures consisting of an alpha and beta subunit. The alpha subunit binds to the cargo protein through the NLS sequence on the cargo protein. The beta subunit interacts with the nucleoporin proteins (Nups) in the
These interactions are shown in Figure \(\PageIndex{21}\).
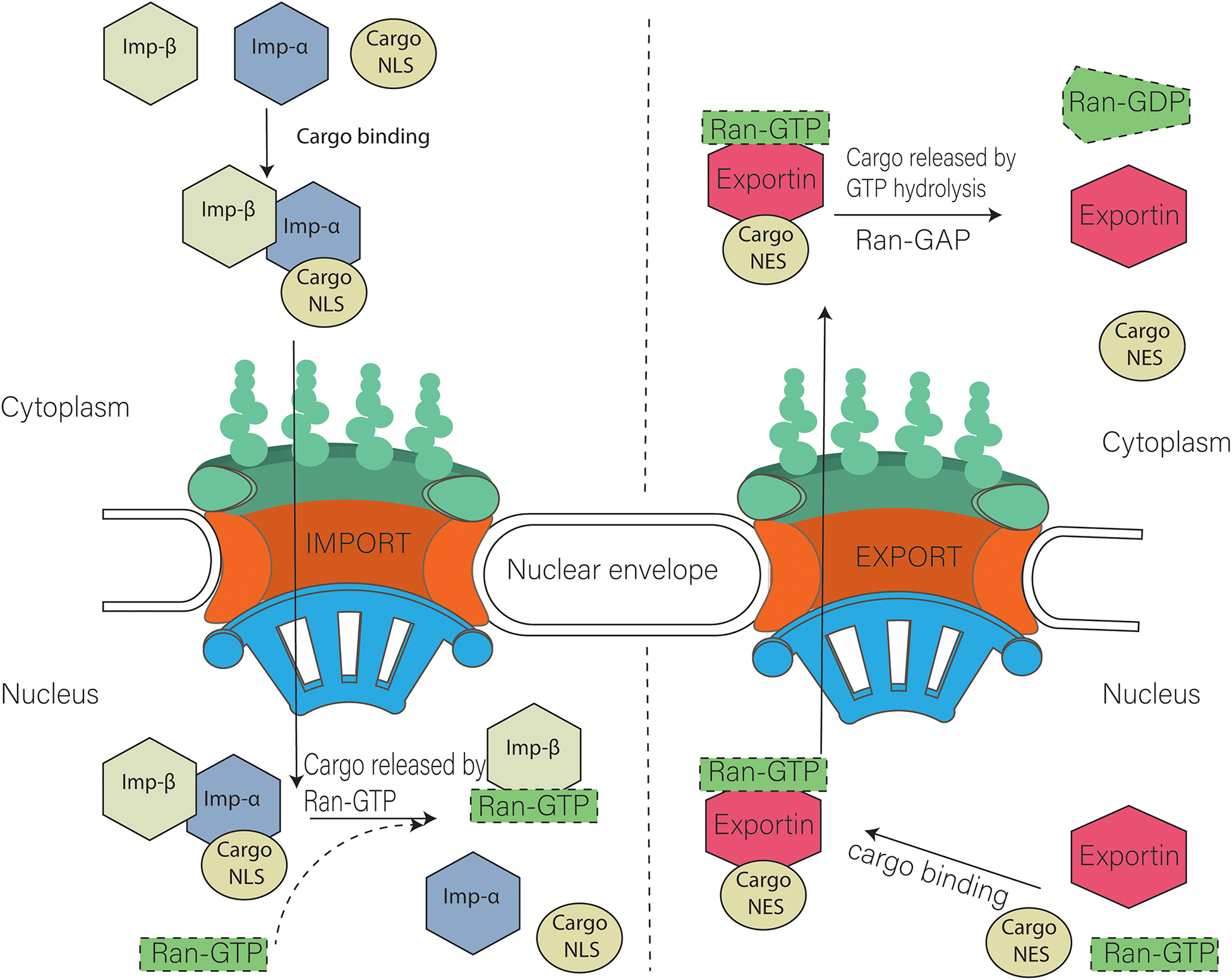
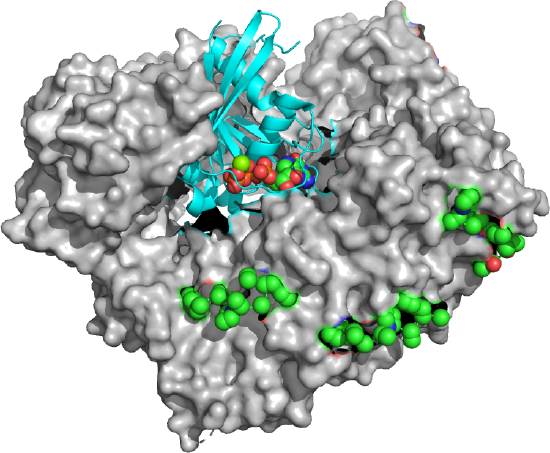
Figure \(\PageIndex{22}\) shows an interactive iCn3D model of the Delpi electrostatic surface potential map of the putative NLS from the carboxy-terminal of a cargo protein, the W protein of the Hendra virus (4M2T)which is imported into the nucleus.
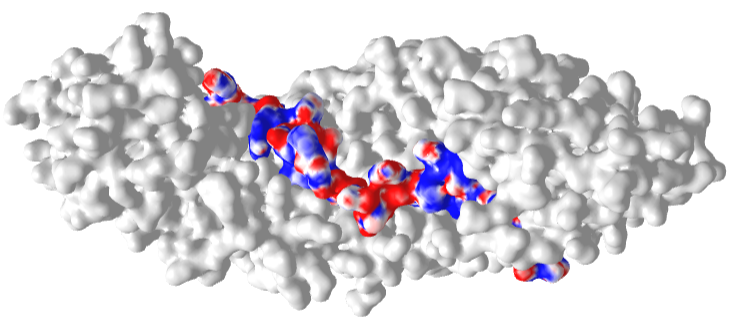
The virus derives from bats and has recently emerged. The peptide sequence containing the NLS has the following sequence: 419 CLGRRVVQPGMFADYPPTKKARVLLR 444. The red surface indicates the negative potential and the blue positive potential, which is associated with the Lys and Arg side chains in the consensus sequence. It is shown bound to the human importin-α3 subunit (molecular surface shown in white), which binds the NLS sequence with high affinity.
An astute reader might ask why GTP stays bound and is not cleaved into GDP in the nucleus by the intrinsic GTPase activity of Ran. It turns out there is yet another protein found only in the cytoplasm (i.e. it doesn't have a nuclear import signal), which binds to Ran:GTP and promotes GTP → GDP exchange. So oddly, hydrolysis of a nucleotide triphosphate (GTP, not ATP as in the case of most transporters discussed above) is required for nuclear import. A different but very fascinating mechanism!
The cargo complexes described above must still pass through the nuclear pore. In section 11.3 we discussed the structure of the nuclear pore which consists of many nucleoporin proteins (NUPs). The inner ring is called the FG Nups layer. The disordered parts of the inner ring Nups have repetitive sequences enriched in phenylalanine and glycine (hence the name FG Nups) and stick out into the central pore. The FG repeats can take multiple forms including Phe-Gly (FG), Gly-Leu-Phe-Gly (GLFG), or Phe-any-Phe-Gly (FXFG). These are intrinsically disordered proteins and the disordered regions in the pore act as a filter allowing certain molecules to pass and excluding those greater with molecular weights greater than 40K. Large molecules complexed to importins/exportins move through the pore. This disordered mesh prevents passive diffusion of molecules through it but allows protein complexes with cargo:exportin/importin complex through. It's a bit like electrophoresis of small protein complexes through the pores of an acrylamide or agarose gel polymerized matrix, only without the "push" of an electric field. Presumably, the FG-Nups make transient hydrophobic (induced-dipole:induced dipole) interactions between the FGs on the Nups and the nuclear transport receptors.
Figure \(\PageIndex{23}\) shows an export complex (5XOJ) from yeast of Ran (cyan with bound GTP in spacefill) bound to an exportin (Xpo1p, white surface) and 3 Nup42p peptides containing SxFG/PxFG repeats (spacefill, side chains, which are mostly nonpolar). To bind 3 Nup peptides, the exportin Xpo1 also contains repeating binding sites on domains called HEAT repeats 14–20 of Xpo1p, The exportin Xpo1p is shaped like a toroid with 21 HEAT repeat domains that have two antiparallel sheets with connecting loops of different sizes.
Some believe the unfolded mesh of FGs in the core might condense on the binding of motifs on importins/exportins or through FG:FG domain associations. But what is the basis for importins/exportins interactions with the FG structures? To study it in more detail Fragasso et al designed and made an artificial FG-Nup that binds to an importin transport receptor Kap95 that interacts with a cargo protein with a nuclear localization sequence (NLS) in cargo proteins. They attached them to the inside of solid-state nanopores and demonstrated the fast movement of Kap95 through the derivatized nanopore while a control "non cargo" without a NLS, BSA) was blocked. Underivatized nanometer-sized pores are made in a silicon nitride synthetic membrane (SiN) by using an ion or electron beam to tune the size of the hole.
Figure \(\PageIndex{24}\) (top image) shows colored code structures of the inner ring (top of figure) snapshots of different yeast GLFG NUPs. These are named after their Gly-Leu-Phe-Gly repeating motifs and are especially cohesive. The GLFG-Nups, shown in red, are mostly found in the inner ring compared to FxFG/FG-Nups shown in green.

The bottom part of Figure \(\PageIndex{24}\) shows snapshots of molecular dynamics simulations of various yeast GLFG-Nups compared to the NupX synthetic one. Three of the yeast GLFG Nups and the synthetic NupX show a compact and extended domain Each has a collapsed cohesive domain at one end (characterized by a low charge to hydrophobic amino acid residue ratio (C/H ), lots of alternating FG and GLFG repeats, with light green showing non-FG/GLFG/charged residues ) and an extended domain at the other end (high ratio of charged/hydrophobic amino acids, no FG repeats, pink-red showing non-FG/GLFG/charged residues). Bright green shows the FG repeats, red the GLFG repeats, and white the charged groups.
Figure \(\PageIndex{25}\) shows the pore in the silicon nitride synthetic membrane (SiN) membrane before (left) and after derivitization with NupX.

Figure \(\PageIndex{26}\) shows derivatized pores in the SiN membrane of increasing size. In the smallest particle, the NupXs don't fit and are expelled from the pore. In the 30 nm particle, the NupX cohesive domains plug the hole. In the 45 nm pore, a hole appears.

Figure \(\PageIndex{26}\): Derivatized pores in the silicon nitride (SiN) membrane of increasing size
Lastly, Figure \(\PageIndex{27}\) shows snapshots of 30 nm pores lines with NupXs. In panel c, an importin, Kap95 (spheres with orange binding spots) is shown forming transient interaction with and translocating through the NupX-lined hole. BSA (sphere without binding sites) may interact weakly but does not translocate through the pore.
