
2.5: Solubility in an aqueous world - The Hydrophobic Effect
- Page ID
- 69576


Introduction
Many biomolecules such as triacylglycerols, cholesterol esters, and waxes are nonpolar. Other biomolecules such as proteins and many lipids have both polar and nonpolar parts. We know from experience that oil floats on the surface of water, showing that it is less dense than water and that it doesn't dissolve in water. You have also probably performed liquid-liquid extractions in chemistry labs in which you utilized the solubility properties of nonpolar molecules to extract them from a mixture in water to a more nonpolar phase such as octanol or chloroform. To understand the stability of biomolecules that contain nonpolar parts in aqueous solutions, we need to understand not only noncovalent interactions of the molecules with water (which we explored in Chapter 2.4) but also the thermodynamics of their molecular interactions in aqueous environments.
We have been taught and internalized the notion that "like-dissolves like". We anthropomorphize molecules to say nonpolar molecules "like" to be in nonpolar environments. We can rationalize solubility properties by examining the noncovalent attractive and repulsive interactions of a molecule in an aqueous solution but when we do so we are usually focusing on enthalpic contributions to stability. What about entropy? We should consider net changes in noncovalent solute:solute, solute:solvent, and solvent:solvent interactions, as well as their thermodynamic contributions to overall stability. When we consider the thermodynamics of the solubility of molecules in water, we need to determine the ΔG, the free energy change, for all processes involved.
The Change in Free Energy (G) and Chemical Potential (μ)
ΔG, the free energy change for a reaction, determines the spontaneity and extent of a chemical or physical reaction. The free energy of a system depends on 3 variables, temperature T, pressure P, and n, the number of moles of each substance. For the latter, think of solute X on two different sides of a permeable membrane. If the concentration of X is the same on each side, as shown in Figure \(\PageIndex{1}\), the system is in equilibrium.

If the system is composed of two different parts, A and B, the system is at equilibrium (ΔG=0) if TA = TB, PA = PB, and the change in the absolute free energy per mole of A is ΔGA/Δn = ΔGB/Δn. More precisely, using simple calculus, we would discuss incremental changes in absolute free energy/mol, dGA/dn for A, which is the chemical potential of A, μA) and dGB/dn (μB) for B. At equilibrium dGA/dn = dGB/dn. We will use the free energy G here but μ later in this section. G then is the absolute free energy/mol (again chemical potential), where G=Go +RTln[A]. From this the equations you used in introductory chemistry (and that we reviewed in Chapter 1.3), the following equation can be written.
\begin{equation}
\begin{array}{l}
\Delta \mathrm{G}=\Delta \mathrm{G}^{0}+\mathrm{RTIn} \mathrm{Qr} \\
\Delta \mathrm{G}=\Delta \mathrm{H}-\mathrm{T} \Delta \mathrm{S} \\
\Delta \mathrm{G}^{0}=\Delta \mathrm{H}^{0}-\mathrm{T} \Delta \mathrm{S}^{0} \\
\Delta \mathrm{G}^{0}=-\mathrm{RTInK} \mathrm{eq}
\end{array}
\end{equation}
Now let's apply this to the chemical equation for the solubility of a given solute in water. If you add either a sparing soluble hydrocarbon (HC) or sodium chloride to water, eventually you reach a point of saturation. For the salt, the water is saturated with dissolved NaCl, and no further increase in NaCl (aq) occurs. For a sparing soluble hydrocarbon, the solution reaches saturation after which phase separation occurs.
Let's add a drop of a slightly soluble hydrocarbon liquid (HCL) into water, as pictured in the diagram below. At t=0, the system is not at equilibrium and some of the HC will transfer from the pure liquid to water, so at time t=0, ΔGTOT < 0. This is illustrated in Figure \(\PageIndex{2}\).

The following equations can be derived.
\begin{equation}
\begin{array}{c}
\Delta \mathrm{G}_{\mathrm{TOT}}=\left(G_{\mathrm{HC}-\mathrm{W}}\right)-\left(G_{\mathrm{HC}-\mathrm{L}}\right)=\mathrm{G}_{\mathrm{HC}-\mathrm{W}}^{0}+R T \ln [\mathrm{HC}]_{\mathrm{W}}-\left(\mathrm{G}_{\mathrm{HC}-\mathrm{L}}^{0}+R T \ln [\mathrm{HC}]_{\mathrm{L}}\right)= \\
\Delta \mathrm{G}_{\mathrm{TOT}}=\left(\mathrm{G}_{\mathrm{HC}-\mathrm{W}}^{0}-\mathrm{G}_{\mathrm{HC}-\mathrm{L}}^{0}\right)+R T \ln \left([\mathrm{HC}]_{\mathrm{W}}-\ln [\mathrm{HC}]_{\mathrm{L}}\right)= \\
\Delta \mathrm{G}_{\mathrm{TOT}}=\Delta \mathrm{G}^{0}+R T \ln \frac{[\mathrm{HC}]_{\mathrm{W}}}{[\mathrm{HC}]_{\mathrm{L}}}
\end{array}
\end{equation}
Now add a bit more complexity to the last example. Add a hydrocarbon x, to a biphasic system of water and octanol as shown in Figure \(\PageIndex{3}\). Shake it vigorously. At equilibrium, x would have "partitioned" between the two mostly immiscible phases.

A simple reaction can be written for this system: X aq ↔ X oct.
Clearly, if X is a hydrocarbon, ΔG < 0 for the reaction written above. Also, ΔGo < 0, since this term is independent of concentration and depends only on the intrinsic stability of X in water in comparison to that of octanol. This simple equation holds:
\begin{equation}
\Delta \mathrm{G}_{\mathrm{TOT}}=\left(\mathrm{G}_{\mathrm{X}-\mathrm{oct}}^{0}-\mathrm{G}_{\mathrm{X}-\mathrm{w}}^{0}\right)+R T \ln \frac{[\mathrm{X}]_{\mathrm{oct}}}{[\mathrm{X}]_{\mathrm{w}}}=\Delta \mathrm{G}^{0}+R T \ln \frac{[\mathrm{X}]_{\mathrm{oct}}}{[\mathrm{X}]_{\mathrm{w}}}
\end{equation}
At equilibrium, ΔG0=0 and the equation can be rewritten as:
\begin{equation}
\Delta \mathrm{G}^{0}=-R T \ln \frac{[\mathrm{X}]_{\mathrm{oct}}}{[\mathrm{X}]_{\mathrm{w}}}=-\mathrm{RTlnK}_{\mathrm{part}}
\end{equation}
where Kpart is the equilibrium partition coefficient for X in octanol and water. This can readily be determined in the lab. Just shake a separatory flask with a biphasic system of octanol and water after injecting a bit of X. Then separate the layers and determine the concentration of x in each phase. Plug these numbers into the last equation. You should be able to predict the sign and relative magnitude of ΔGo since it does not depend on concentration, but only on the intrinsic stability of the molecules in the different environments. Kpart values are often determined for drugs since they often must diffuse across cell membranes to move into the cytoplasm where they can act. Drugs hence must have a reasonable Kpart to pass through the membrane but not so high that they are insoluble.
Introduction to the Hydrophobic Effect
Now let's ask this question: What are the enthalpic and entropic contributions to the ΔG for the interaction of a nonpolar molecule HC with water? For this section, we will replace ΔG with Δμ (the change in chemical potential but we will use these terms interchangeably). Likewise, we will use this equation: Δμo = ΔHo - T ΔSo.
Also instead of framing the reaction as the dissolution of an organic molecule in water, we will frame is as the transfer of a hydrocarbon X from an aqueous solution to the pure hydrocarbon liquid (HC) or
\begin{equation}
\mathrm{X}(\mathrm{aq}) \leftrightarrow \mathrm{X}(\mathrm{HC})
\end{equation}
Figure \(\PageIndex{4}\) shows the standard free energies of transfer of a hydrocarbon X from an aqueous solution to a pure liquid hydrocarbon (HC), X (aq) ↔ X (HC). where
\begin{equation}
\Delta \mu^{\circ}=\mu^{\circ} x(H C)-\mu^{\circ} x(a q)
\end{equation}
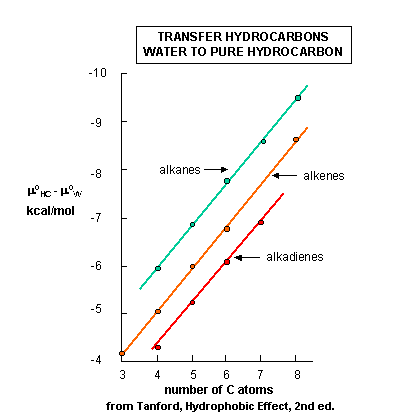
Δμo is less than 0 since transfer back to the pure HC is favored from a stability perspective. In each graph, Δμo is less than 0, and the value of Δμo decreases (gets more negative as you go up the y axis which shows increasingly negative values of Δμo) in a linear fashion with increasing numbers of carbon atoms in the alkyl chain. Notice the lines are unbelievably straight and parallel. Nature is speaking to us in these figures. By determining the surface area of the hydrocarbon molecules and the decrease in Δμo with each added CH2 (methylene group), one can calculate that the Δμo decreases by 25 cal/Å2 (105 J/Å2), per methylene added.
We expected that Δμo for the transfer of X to a pure liquid HC would be negative. We could get more information if we would determine both the entropic and enthalpic contributions. Such data is presented in the table below, which shows the transfer of short, single-chain alcohol X (an amphiphile with a polar head and a longer nonpolar "tail") from the pure liquid alcohol (ROH) to water (the opposite of the previous figures.)
\begin{equation}
X(R O H) \leftrightarrow X(W)
\end{equation}
Thermodynamic Parameters for Transfer of Aliphatic Alcohol X from the Pure Liquid to Water at 25oC (enthalpy determined by calorimetry)
| alcohol X | μw0-μ ROH0 kcal/mol (kJ/mol) | Hw0-H ROH0 kcal/mol (kJ/mol) | Sw0-S ROH0 cal/deg mol (J/deg mol) |
(Cp)w0-(Cp)ROH0 cal/deg mol |
| ethanol | 0.760 (3.18) | -2.43 (-10.2) | -10.7 (-44.8) | 39 (163) |
| n-propanol | 1.58 (6.61) | -2.42 (-10.2) | -13.4 (-56.1) | 56 (234) |
| n-butanol | 2.4 (10) | -2.25 (-9.41) | -15.6 (-65.3) | 72 (301) |
| n-pentanol (solubility 22g/L H2O) |
3.22 (13.5) | -1.87 (-7.82) | -17.1 (-71.5) | 84 (352) |
We expect the Δμo to be increasingly positive as the chain length gets longer and their solubilities in water become increasingly disfavored. What is perplexing about this data is not that the transfer of these ROHs to water is disfavored, but that transfer is enthalpically favored (negative ΔH0). This seems to be counterintuitive since it goes against the adage that "like dissolves like" as was discussed earlier. From an enthalpic point of view, the amphiphiles prefer (albeit marginally) to be in water. What makes this reaction disfavored is entropy. The data shows that the nonpolar molecule would prefer not to be in the water because it is disfavored entropically.
At first glance, you might guess that the entropy should favor the movement of ROHs into water since they could access a larger volume to access and have greater freedom of motion. Hence here are more possible microstates for the ROH in water. However, this is only part of the process. What we haven't considered is the entropy of the water. To place a hydrocarbon in water, a literal cavity in the water must be created that will accommodate it. Creation of this more ordered cavity must be entropically disfavored (again because the process proceeds to a state with fewer microstates and lower positional entropy).
For the reverse process, transferring the hydrocarbon from water to the pure liquid will dissipate the cavity, and lead to more available microstates for the released solvent, bulk water. It is this entropic contribution that favors the movement of a hydrocarbon from water to the pure hydrocarbon lipid. This "hydrophobic effect" is the main thermodynamic drive to move organic molecules out of water.
Image this scenario. When you place a hydrocarbon group into water, water seeks (admittedly an anthropomorphic term) to maintain its hydrogen bonding. Hence it is forced into a more ordered structure around the HC to maintain its H-bonding, characterized by fewer microstates. We will explore the hydrophobic effect in greater detail in a future chapter.
How can we explain the favorable enthalpic contribution on placing a nonpolar molecule into water? Again, this goes against our adage of "like dissolves like". The negative ΔH suggests interactions among all the participants are more favorable when the nonpolar group is in water. One source of such interactions could be the highly structured water in the "cage" that surrounds the nonpolar molecule. If it were more structured than bulk water, hence more "ice-like" in nature, then the formation of these extra H-bonds would contribute to the negative enthalpy change. When the nonpolar molecule is removed from the water, a process which proceeds with a positive ΔH, the cage of "ice-like" water would "melt", which, like the melting of ice, is not favored enthalpically, as heat must be added. Heat energy must be supplied to break the H-bonds as ice changes state to liquid water. This molecular model to understand the thermodynamic data might yet be a simplistic model, but for time being, let's use it.