
8.3: Isolating or Detecting a Specific Sequence by PCR
- Page ID
- 4109

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Components of the PCR Reaction
The Polymerase Chain Reaction (PCR) is a method of DNA replication that is performed in a test tube (i.e. in vitro). Here “polymerase” refers to a DNA polymerase enzyme extracted and purified from bacteria, and “chain reaction” refers to the ability of this technique produce millions of copies of a DNA molecule, by using each newly replicated double helix as a template to synthesize two new DNA double helices. PCR is therefore a very efficient method of amplifying DNA.
Besides its ability to make large amounts of DNA, there is a second characteristic of PCR that makes it extremely useful. Recall that most DNA polymerases can only add nucleotides to the end of an existing strand of DNA, and therefore require a primer to initiate the process of replication. For PCR, chemically synthesized primers of about 20 nucleotides are used. In an ideal PCR, primers only hybridize to their exact complementary sequence on the template strand (Figure \(\PageIndex{3}\)).

The experimenter can therefore control exactly what region of a DNA template is amplified by controlling the sequence of the primers used in the reaction.
To conduct a PCR amplification, an experimenter combines in a small, thin-walled tube (Figure \(\PageIndex{4}\)), all of the necessary components for DNA replication, including DNA polymerase and solutions containing nucleotides (dATP, dCTP, dGTP, dTTP), a DNA template, DNA primers, a pH buffer, and ions (e.g. Mg2+) required by the polymerase. Successful PCR reactions have been conducted using only a single DNA molecule as a template, but in practice, most PCR reactions contain many thousands of template molecules. The template DNA (e.g. total genomic DNA) has usually already been purified from cells or tissues using the techniques described above. However, in some situations it is possible to put whole cells directly in a PCR reaction for use as a template.
An essential aspect of PCR is thermal-cycling, meaning the exposure of the reaction to a series of precisely defined temperatures (Figure \(\PageIndex{5}\)). The reaction mixture is first heated to 95°C. This causes the hydrogen bonds between the strands of the template DNA molecules to melt, or denature. This produces two single-stranded DNA molecules from each double helix (Figure \(\PageIndex{6}\)). In the next step (annealing), the mixture is cooled to 45-65°C. The exact temperature depends on the primer sequence used and the objectives of the experiment. This allows the formation of double stranded helices between complementary DNA molecules, including the annealing of primers to the template. In the final step (extension) the mixture is heated to 72°C. This is the temperature at which the particular DNA polymerase used in PCR is most active. During extension, the new DNA strand is synthesized, starting from the 3' end of the primer, along the length of the template strand. The entire PCR process is very quick, with each temperature phase usually lasting 30 seconds or less. Each cycle of three temperatures (denaturation, annealing, extension) is usually repeated about 30 times, amplifying the target region approximately 230-fold. Notice from the figure that most of the newly synthesized strands in PCR begin and end with sequences either identical to or complementary to the primer sequences; although a few strands are longer than this, they are in such a small minority that they can almost always be ignored.
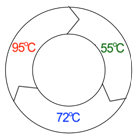
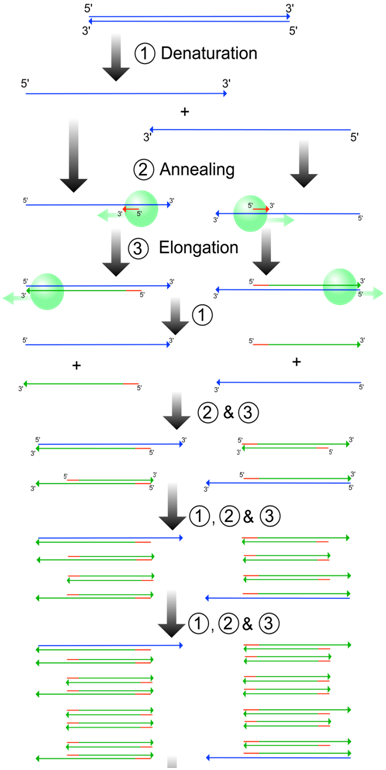
Figure \(\PageIndex{6}\): PCR with the three phases of the thermalcycle numbered. The template strand (blue) is replicated from primers (red), with newly synthesized strands in green. The green strands flanked by two primer binding sites will increase in abundance exponentially through successive PCR cycles. (Wikipedia-madprime-GFDL)
The earliest PCR reactions used a polymerase from E. coli. Because the high temperature of the denaturation step destroyed the enzyme, new polymerase had to be added after each cycle. To overcome this, researchers identified thermostable DNA polymerases such as Taq DNA pol, from Thermus acquaticus, a thermophilic bacterium that lives in hot springs. Taq, and similar thermostable polymerases from other hot environments, are able to remain functional in the repeated cycles of amplification. Taq polymerase cannot usually amplify fragments longer than about 3kbp, but under some specialized conditions, PCR can amplify fragments up to approximately 10kbp. Other polymerases, either by themselves or in combination with Taq, are used to increase the length of amplified fragments or to increase the fidelity of the replication.
After completion of the thermalcycling (amplification), an aliquot from the PCR reaction is usually loaded onto an electrophoretic gel (described below) to determine whether a DNA fragment of the expected length was successfully amplified or not. Usually, the original template DNA will be so dilute that it will not be visible on the gel, only the amplified PCR product. The presence of a sharp band of the expected length indicates that PCR was able to amplify its target. If the purpose of the PCR was to test for the presence of a particular template sequence, this is the end of the experiment. Otherwise, the remaining PCR product can be used as starting material for a variety of other techniques such as sequencing or cloning.
PCR is very sensitive (meaning it can amplify very small starting amounts of DNA), and specific (meaning it can amplify only the target sequence from a mixture of many DNA sequences). This made PCR the perfect tool to test whether genetically modified corn was present in consumer products on supermarket shelves. Although currently (2013) 85% of corn in the United States is genetically modified, and contains genes that government regulators have approved for human consumption, back in 2000, environmental groups showed that a strain of genetically modified corn, which had only been approved for use as animal feed, had been mixed in with corn used in producing human food, like taco shells.
To do this, the groups purchased taco shells from stores in the Washington DC area, extracted DNA from the taco shells and used it as a template in a PCR reaction with primers specific to the unauthorized gene (Cry9C). Their suspicions were confirmed when they ran this PCR product on an agarose gel and saw a band of expected size. The PCR test was able to detect one transgenic kernel in a whole bushel of corn (1 per 100,000). The company (Aventis) that sold the transgenic seed to farmers had to pay for the destruction of large amounts of corn, and was the target of a class action law-suit by angry consumers who claimed they had been made sick by the taco shells. While no legitimate cases of harm were ever proven, and the plaintiffs were awarded $9 million, of which $3 million went to the legal fees, and the remainder of the judgment went to the consumers in the form of coupons for taco shells. The affair damaged the company, and exposed a weakness in the way the genetically modified crops were handled in the United States at the time.