
S2018_Lecture18_Reading
- Page ID
- 11507
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Nucleic acids
There are two types of nucleic acids in biology: DNA and RNA. DNA carries the heritable genetic information of the cell and is composed of two antiparallel strands of nucleotides arranged in a helical structure. Each nucleotide subunit is composed of a pentose sugar (deoxyribose), a nitrogenous base, and a phosphate group. The two strands associate via hydrogen bonds between chemically complementary nitrogenous bases. Interactions known as "base stacking" interactions also help stabilize the double helix. By contrast to DNA, RNA can be either be single stranded, or double stranded. It too is composed of a pentose sugar (ribose), a nitrogenous base, and a phosphate group. RNA is a molecule of may tricks. It is involved in protein synthesis as a messenger, regulator, and catalyst of the process. RNA is also involved in various other cellular regulatory processes and helps to catalyze some key reactions (more on this later). With respect to RNA, in this course we are primarily interested in (a) knowing the basic molecular structure of RNA and what distinguishes it from DNA, (b) understanding the basic chemistry of RNA synthesis that occurs during a process called transcription, (c) appreciating the various roles that RNA can have in the cell, and (d) learning the major types of RNA that you will encounter most frequently (i.e. mRNA, rRNA, tRNA, miRNA etc.) and associating them with the processes they are involved with. In this module we focus primarily on the chemical structures of DNA and RNA and how they can be distinguished from one another.
Nucleotide structure
The two main types of nucleic acids are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). DNA and RNA are made up of monomers known as nucleotides. Individual nucleotides condense with one another to form a nucleic acid polymer. Each nucleotide is made up of three components: a nitrogenous base (for which there are five different types), a pentose sugar, and a phosphate group. These are depicted below. The main difference between these two types of nucleic acids is the presence or absence of a hydroxyl group at the C2 position, also called the 2' position (read "two prime"), of the pentose (see Figure 1 legend and section on the pentose sugar for more on carbon numbering). RNA has a hydroxyl functional group at that 2' position of the pentose sugar; the sugar is called ribose, hence the name ribonucleic acid. By contrast, DNA lacks the hydroxyl group at that position, hence the name, "deoxy" ribonucleic acid. DNA has a hydrogen atom at the 2' position.

The nitrogenous base
The nitrogenous bases of nucleotides are organic molecules and are so named because they contain carbon and nitrogen. They are bases because they contain an amino group that has the potential of binding an extra hydrogen, and thus acting as a base by decreasing the hydrogen ion concentration in the local environment. Each nucleotide in DNA contains one of four possible nitrogenous bases: adenine (A), guanine (G), cytosine (C), and thymine (T). By contrast, RNA contains adenine (A), guanine (G) cytosine (C), and uracil (U) instead of thymine (T).
Adenine and guanine are classified as purines. The primary distinguishing structural feature of a purine is double carbon-nitrogen ring. Cytosine, thymine, and uracil are classified as pyrimidines. These are structurally distinguished by a single carbon-nitrogen ring. You will be expected to recognize that each of these ring structures is decorated by functional groups that may be involved in a variety of chemistries and interactions.
Note: practice
Take a moment to review the nitrogenous bases in Figure 1. Identify functional groups as described in class. For each functional group identified, describe what type of chemistry you expect it to be involved in. Try to identify whether the functional group can act as either a hydrogen bond donor, acceptor, or both?
The pentose sugar
The pentose sugar contains five carbon atoms. Each carbon atom of the sugar molecule are numbered as 1′, 2′, 3′, 4′, and 5′ (1′ is read as “one prime”). The two main functional groups that are attached to the sugar are often named in reference to the carbon to whch they are bound. For example, the phosphate residue is attached to the 5′ carbon of the sugar and the hydroxyl group is attached to the 3′ carbon of the sugar. We will often use the carbon number to refer to functional groups on nucleotides so be very familiar with the structure of the pentose sugar.
The pentose sugar in DNA is called deoxyribose, and in RNA, the sugar is ribose. The difference between the sugars is the presence of the hydroxyl group on the 2' carbon of the ribose and its absence on the 2' carbon of the deoxyribose. You can, therefore, determine if you are looking at a DNA or RNA nucleotide by the presence or absence of the hydroxyl group on the 2' carbon atom—you will likely be asked to do so on numerous occasions, including exams.
The phosphate group
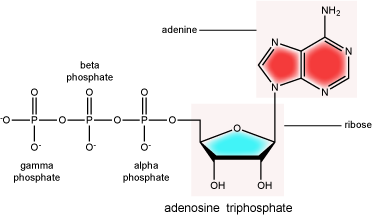
There can be anywhere between one and three phosphate groups bound to the 5' carbon of the sugar. When one phosphate is bound, the nucleotide is referred to as a Nucleotide MonoPhosphate (NMP). If two phosphates are bound the nucleotide is referred to as Nucleotide DiPhosphate (NDP). When three phosphates are bound to the nucleotide it is referred to as a Nucleotide TriPhosphate (NTP). The phosphoanhydride bonds between that link the phosphate groups to each other have specific chemical properties that make them good for various biological functions. The hydrolysis of the bonds between the phosphate groups is thermodynamically exergonic in biological conditions; nature has evolved numerous mechanisms to couple this negative change in free energy to help drive many reactions in the cell. Figure 2 shows the structure of the nucleotide triphosphate Adenosine Triphosphate, ATP, that we will discuss in greater detail in other chapters.
Note: "high-energy" bonds
The term "high-energy bond" is used A LOT in biology. This term is, however, a verbal shortcuts that can cause some confusion. The term refers to the amount of negative free energy associated with the hydrolysis of the bond in question. The water (or other equivalent reaction partner) is an important contributor to the energy calculus. In ATP, for instance, simply "breaking" a phosphoanhydride bond - say with imaginary molecular tweezers - by pulling off a phosphate would not be energetically favorable. We must, therefore, be careful not to say that breaking bonds in ATP is energetically favorable or that it "releases energy". Rather, we should be more specific, noting that they hydrolysis of the bond is energetically favorable. Some of this common misconception is tied to, in our opinion, the use of the term "high energy bonds". While in Bis2a we have tried to minimize the use of the vernacular "high energy" when referring to bonds, trying instead to describe biochemical reactions by using more specific terms, as students of biology you will no doubt encounter the potentially misleading - though admittedly useful - short cut "high energy bond" as you continue in your studies. So, keep the above in mind when you are reading or listening to various discussions in biology. Heck, use the term yourself. Just make sure that you really understand what it refers to.
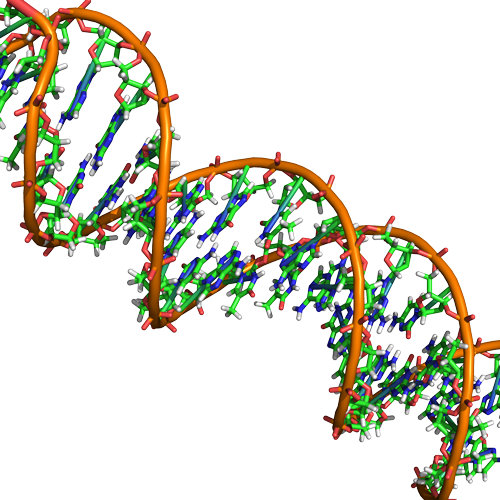
Double helix structure of DNA
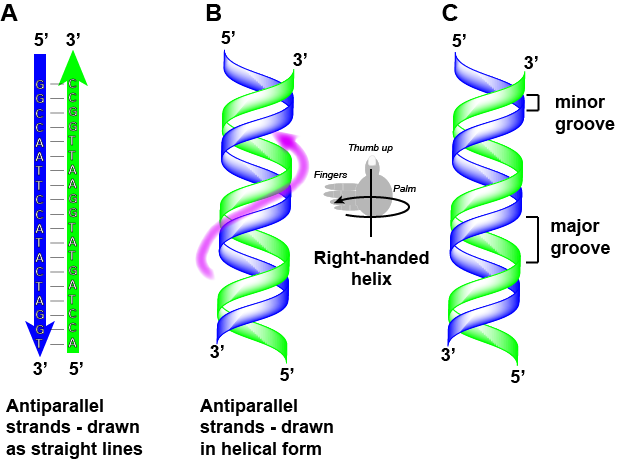
DNA has a double helix structure (shown below) created by two strands of covalently linked nucleotide subunits. The sugar and phosphate groups of each strand of nucleotides are positioned on the outside of the helix, forming the backbone of the DNA (highlighted by the orange ribbons in Figure 3). The two strands of the helix run in opposite directions, meaning that the 5′ carbon end of one strand will face the 3′ carbon end of its matching strand (See Figures 4 and 5). We referred to this orientation of the two strands as antiparallel. Note too that phosphate groups are depicted in Figure 3 as orange and red "sticks" protruding from the ribbon. The phosphates are negatively charged at physiological pHs and therefore give the backbone of the DNA a strong local negatively charged character. By contrast, the nitrogenous bases are stacked in the interior of the helix (these are depicted as green, blue, red, and white sticks in Figure 3). Pairs of nucleotides interact with one another through specific hydrogen bonds (shown in Figure 5). Each pair of separated from the next base pair in the ladder by 0.34 nm and this close stacking and planar orientation gives rise to energetically favorable base-stacking interactions. The specific chemistry associated with these interactions is beyond the content of Bis2a but is described in more detail here for the curious or more advanced students. We do expect, however, that students are aware that the stacking of the nitrogenous bases contributes to the stability of the double helix and defer to your upper-division genetics and organic chemistry instructors to fill in the chemical details.
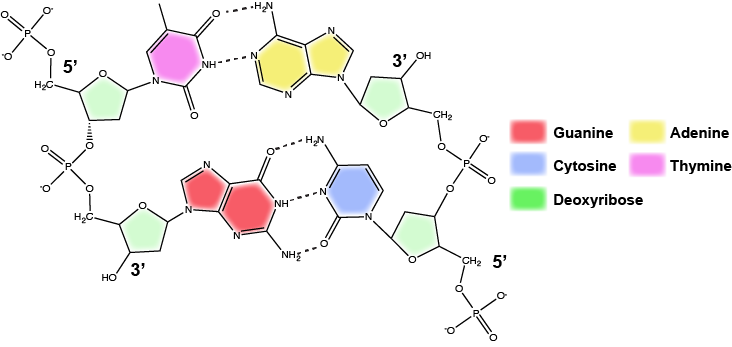
In a double helix, certain combinations of base pairing are chemically more favored than others based on the types and locations of functional groups on the nitrogenous bases of each nucleotide. In biology we find that:
Adenine (A) is chemically complementary with thymidine (T) (A pairs with T)
and
Guanine (G) is chemically complementary with cytosine (C) (G pairs with C).
We often refer to this pattern as "base complementarity" and say that the antiparallel strands are complementary to each other. For example, if the sequence of one strand is of DNA is 5'-AATTGGCC-3', the complementary strand would have the sequence 5'-GGCCAATT-3'.
We sometimes choose to represent complementary double-helical structures in text by stacking the complementary strands on top of on another as follows:
5' - GGCCAATTCCATACTAGGT - 3'
3' - CCGGTTAAGGTATGATCCA - 5'
Note that each strand has its 5' and 3' ends labeled and that if one were to walk along each strand starting from the 5' end to the 3' end that the direction of travel would be opposite the other for each strand; the strands are antiparallel. We commonly say things like "running 5-prime to 3-prime" or "synthesized 5-prime to 3-prime" to refer to the direction we are reading a sequence or the direction of synthesis. Start getting yourself accustomed to this nomenclature.
Functions and roles of nucleotides and nucleic acids to look out for in Bis2a
In addition to their structural roles in DNA and RNA, nucleotides such as ATP and GTP also serve as mobile energy carriers for the cell. Some students are surprised when they learn to appreciate that the ATP and GTP molecules we discuss in the context of bioenergetics are the same as those involved in the formation of nucleic acids. We will cover this in more detail when we discuss DNA and RNA synthesis reactions. Nucleotides also play important roles as co-factors in many enzymatically catalyzed reactions.
Nucleic acids, RNA in particular, play a variety of roles in in cellular process besides being information storage molecules. Some of the roles that you should keep an eye out for as we progress through the course include: (a) Riboprotein complexes - RNA-Protein complexes in which the RNA serves both catalytic and structural roles. Examples of such complexes include, ribosomes (rRNA), RNases, splicesosome complexes, and telomerase. (b) Information storage and transfer roles. These roles include molecules like DNA, messenger RNA (mRNA), transfer RNA (tRNA). (c) Regulatory roles. Examples of these include various non-coding (ncRNA). Wikipedia has a comprehensive summary of the different types of known RNA molecules that we recommend browsing to get a better sense of the great functional diversity of these molecules.
Genomes as organismal blueprints
A genome, not to be confused with a gnome, is an organism's complete collection of heritable information stored in DNA. Differences in information content help to explain the diversity of life we see all around us. Changes to the information encoded in the genome are the primary drivers of the phenotypic diversity we see (and some we can't) around us that are filtered by natural selection, and they are thus the drivers of evolution. This leads to questions. If every cell in a multicellular organism contains the same sequence of DNA, how can there be different cell types (e.g., how can a cell in a liver be so different from a cell in the brain if they both carry the same DNA)? How do we read the information? How do we interpret what we read? How do we understand how all of the "parts" we identify in the genome functionally interrelate? How is all of this related to the expression of traits? How do changes in the genome lead to changes in traits?
Determining a genome sequence
The information encoded in genomes provides important data for understanding life, its functions, its diversity, and its evolution. Therefore, it stands to reason that a reasonable place to begin studies in biology would be to read the information content encoded in the genome(s) in question. A good starting point is to determine the sequence of nucleotides (A, G, C, T) and their organization into one or more independently replicating units of DNA (e.g., think chromosomes and/or plasmids ). For 30+ years after the discovery that DNA is the hereditary material, this was a daunting proposition. In the late 1980s, however, the advent of semi-automated tools for DNA sequencing were pioneered, and this began a revolution that has dramatically changed how we approach the study of life. Twenty years later, in the mid-2000s, we entered a period of accelerated technological progress in which advances in materials sciences (particularly, advances in our ability to make things on a very small scale), optics, electrical and computer engineering, bioengineering, and computer sciences have all converged to bring us dramatic increases in our capacity to sequence DNA and correspondingly dramatic decreases in the cost of numerous advances in our ability to sequence DNA. A famous example to illustrate this point is to compare the changes in cost to sequence the human genome. The first draft of the human genome took nearly 15 years and $3 billion dollars to complete. Today, 10's of human genomes can be sequenced in a single day on a single instrument at a cost of less than $1000 each (the cost and time continue to decrease). Today, companies like Illumina, Pacific Biosciences, Oxford Nanopore, and others offer competing technologies that are driving down the cost and increasing the volume, quality, speed, and portability of DNA sequencing.
One of the very exciting elements of the DNA sequencing revolution is that it has required and continues to require contributions from biologists, chemists, materials scientists, electrical engineers, mechanical engineers, computer scientists and programmers, mathematicians and statisticians, product developers, and many other technical experts. The potential applications and implications of unlocking barriers to DNA sequencing have also engaged investors, business people, product developers, entrepreneurs, ethicists, policy makers, and many others to pursue new opportunities and to think about how to best and most responsibly use this growing technology.
The technological advances in genome sequencing have resulted in a virtual flood of complete genome sequences being determined and deposited into publicly available databases. You can find many of them at the National Center for Biotechnology Information. The number of available , completely sequenced genomes numbers in the tens of thousands—over 2,000 eukaryotic genomes, over 600 archaeal genomes, and nearly 12,000 bacterial genomes at the time of this writing. Tens of thousands of more genome sequencing projects are in progress. With this many genome sequences available—or soon to be available—we can start asking many questions about what we see in these genomes. What patterns are common to all genomes? How many genes are encoded in genomes? How are these organized? How many different types of features can we find? What do the features that we find do? How different are the genomes from one another? Is there evidence that can tell us how genomes evolve? Let's briefly examine a few of these questions.
Diversity of genomes
Diversity of sizes, number of genes, and chromosomes
Let's start by examining the range of genome sizes. In the table below, we see a sampling of genomes from the database. We can see that the genomes of free living organisms range tremendously in size. The smallest known genome is encoded in 580,000 base pairs while the largest is 150 billion base pairs—for reference, recall that the human genome is 3.2 billion base pairs. That's a huge range of sizes. Similar disparities in the number of genes also exist.
Table 1. This table shows some genome data for various organisms. 2n = diploid number. Attribution: Marc T. Facciotti (own work—reproduced from http://book.bionumbers.org/how-big-are-genomes/)

Examining Table 1 also reveals that some organisms carry with them more than one chromosome. Some genomes are also polyploid, meaning that they maintain multiple copies of similar but not identical (homologous) copies of each chromosome. A diploid organism carries in its genome two homologous copies (usually one from Mom and one from Dad) of each chromosome. Humans are diploid. Our somatic cells carry 2 homologous copies of 23 chromosomes. We received 23 copies of individual chromosomes from our mother and 23 copies from our father, for a total of 46. Some plants have higher ploidy. For example, a plant with four homologous copies of each chromosome is termed tetraploid. An organism with a single copy of each chromosome is termed haploid.
Structure of genomes
Table 1 also provides clues to other points of interest. For instance, if we compare the pufferfish genome to the chimpanzee genome, we note that they encode roughly the same number of genes (19,000), but they do so on dramatically differently sized genomes—400 million base pairs versus 3.3 billion base pairs, respectively. That implies that the pufferfish genome must have much less space between its genes than what might be expected to be found in the chimpanzee genome. Indeed, this is the case, and the difference in gene density is not unique to these two genomes. If we look at Figure 1, which attempts to represent a 50-kb part of the human genome, we notice that in addition to the protein-coding regions (indicated in red and pink) that many other so-called "features" can be read from the genome. Many of these elements contain highly repetitive sequences.

Figure 1. This figure shows a 50-kb segment of the human β T-cell receptor locus on chromosome 7. This figure depicts a small region of the human genome and the types of "features" that can be read and decoded in the genome, including, but also in addition to, protein-coding sequences. Red and pink correspond to regions that encode proteins. Other colors represent different types of genomic elements. Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
If we now look at what fraction of the whole human genome each of these types of elements makes up (see Figure 2), we see that protein-coding genes only make up 48 million of the 3.2 billion bases of the haploid genome.

Figure 2. This graph depicts how the many base pairs of DNA in the human haploid genome are distributed between various identifiable features. Note that only a small fraction of the genome is associated directly with protein-coding regions. Attribution: Marc T. Facciotti (own work—reproduced from sources noted in figure)
When we examine the frequency of repeat regions versus protein-coding regions in different species, we note large differences in protein-coding versus non-coding regions.

Figure 3. This figure shows 50-kb segments of different genomes, illustrating the highly variable frequency of repeat versus protein-coding elements in different species.
Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
Suggested discussion
Propose a hypothesis for why you think some genomes might have more or fewer noncoding sequences.
Dynamics of genome structure
Genomes change over time, and numerous different types of events can change their sequence.
1. Mutations are either accumulated during DNA replication or through environmental exposure to chemical mutagens or radiation. These changes typically occur at the level of single nucleotides.
2. Genome rearrangements describe a class of large-scale changes that can occur, and they include the following: (a) deletions—where segments of the chromosome are lost; (b) duplication—where regions of the chromosome are inadvertently duplicated; (c) insertions—the insertion of genetic material (note that sometimes this is acquired from viruses or the environment, and deletion/insertion pairs may happen across chromosomes); (d) inversions—where regions of the genome are flipped within the same chromosome; and (e) translocations—where segments of the chromosome are translocated (moved elsewhere in the chromosome).
These changes happen at different rates, and some are facilitated by the activity of enzyme catalysts (e.g., transposases).
The study of genomes
Comparative genomics
One of the most common things to do with a collection of genome sequences is to compare the sequences of multiple genomes to one another. In general terms, these types of activities fall under the umbrella of a field called comparative genomics.
Comparing the genomes of people who suffer from an inheritable disease to the genomes of people who are not afflicted can help us to uncover the genetic basis for the malady. Comparing the gene content, order, and sequence of related microbes can help us find the genetic basis of why some microbes cause disease while their close cousins are virtually harmless. We can compare genomes to understand how a new species may have evolved. There are many possible analyses! The basis of these analyses is similar: look for differences across multiple genomes and try to associate those differences with different traits or behaviors in those organisms.
Lastly, some people are comparing genome sequences to try to understand the evolutionary history of the organisms. Typically, these types of comparisons result in a graph known as a phylogenetic tree, which is a graphical model of the evolutionary relationship between the various species being compared. This field, not surprisingly, is called phylogenomics.
Metagenomics: who is living somewhere and what are they doing?
In addition to studying the genomes of individual species, the increasingly powerful DNA-sequencing technologies are making it possible to simultaneously sequence the genomes of environmental samples that are inhabited by many different species. This field is called metagenomics. These studies are typically focused on trying to understand what microbial species inhabit different environments. There is great interest in using DNA sequencing to study the populations of microbes in the gut and to watch how the population changes in response to different diets, to see if there is any association between the abundance of different microbes and various diseases, or to look for the presence of pathogens. People are using DNA sequencing of environmental metagenomic samples to explore which microbes inhabit different environments on Earth (from the deep sea, to soil, to air, to hypersaline ponds, to cat feces, to some of the common surfaces we touch every day).
In addition to discovering "who lives where," the sequencing of microbial populations in different environments can also reveal what protein-coding genes are present in an environment. This can give investigators clues into what metabolic activities might be occurring in that environment. In addition to providing important information about what kind of chemistry might be happening in a specific environment, the catalog of genes that is accumulated can also serve as an important resource for the discovery of novel enzymes for applications in biotechnology.