
Lecture 25 and 26: Gene Expression Regulation
( \newcommand{\kernel}{\mathrm{null}\,}\)
Introduction to gene regulation
Regulation is all about decision making. Gene regulation is, therefore, all about understanding how cells make decisions about which genes to turn on, turn off or to tune up or tune down. In the following section we discuss some of the fundamental mechanisms and principles used by cells to regulate gene expression in response to changes in cellular or external factors. This biology is important for understanding how cells adjust changing environments, including how some cells, in multicellular organisms, decide to become specialized for certain functions (e.g. tissues).
Since the subject of regulation is both a very deep and broad topic of study in biology, in Bis2a we don't try to cover every detail - there are simply way too many. Rather, as we have done for all other topics, we try to focus on (a) outlining some of the core logical constructs and questions that you must have when you approach ANY scenario involving regulation, (b) learning some common vocabulary and ubiquitous mechanisms and (c) examining a few concrete examples that illustrate the points made in a and b.
Gene Expression
Introduction
All cells control when and how much each one of its genes are expressed. This simple statement - one that could be derived simply from observing cellular behavior - brings up many questions that we can begin to lay out using our Design Challenge rubric.
Trying to define "gene expression"
The first thing we need to do, however, is to define what it means when we say that a gene is "expressed". If the gene encodes a protein, one might reasonably propose that "expression" of a gene means how much functional protein is made. But what if the gene does not encode a protein, but rather some functional RNA. Then, in this case, "expressed might mean how much of the functional RNA is made. Yet another person might reasonably suggest that "expression" just refers to the initial step in creating a copy of the genomic information. By that definition, one might want to count how many full-length transcripts are being made. Is it the number of end products encoded by the genomic information or is it the number of reads of the information that is important to properly describe "expression". Unfortunately, in practice we often find that the definition depends on the context of the discussion. Keep that in mind. For the sake of making sure that we are talking about the same thing, in Bis2A we'll try to use the term "expression" primarily to describe the creation of the final functional product(s). Depending on the specific case, the final product may be a protein or RNA species.
The design challenge of regulating gene expression
To drive this discussion from a design challenge perspective, we can formally stipulate that the "big problem" we are interested in understating is that of regulating protein abundance in a cell. Problem: The abundance of each functional protein must be regulated. We can then start by posing subproblems:
Let's take a moment, though, first to reload a couple of ideas. The process of gene expression requires multiple steps depending on what the fate of the final product will be. In the case of structural and regulatory RNAs (i.e. tRNA, rRNA, snRNA, etc.) the process requires that a gene be transcribed and that any needed post-transcriptional processing take place. In the case of a protein coding gene, the transcript must also be translated into protein and if required, modifications to the protein must also be made. Of course, both transcription and translation are multi-step processes and most those sub-steps are also potential sites of control.
Some of the subproblems might therefore be:
1. It is reasonable to postulate that there must be some mechanism(s) to regulate the first step of this multi-step process, the initiation of transcription (just getting things started). So, we could state, "we need a mechanism to regulate the initiation of transcription." We could also turn this into a question and ask, "how can the initiation of transcription be accomplished"?
2. We can use similar thinking to state, "we need a mechanism for regulating the end of transcription" or to ask "how is transcription terminated?"
3. Using this convention we can state, "we need to regulate the initiation of translation and the stop of translation".
4. We've talked only about synthesis of protein and RNA. It is quite reasonable to also state, "we need a mechanisms to regulate the degradation of RNA and protein."
Focusing on transcription
In this course we begin by focusing primarily on examining the first couple of problems/questions, the regulation of transcription initiation and termination - from genomic information to a functional RNA, either ready as is (e.g. in the case of a functional RNA) or ready for translation. This allows us to examine some fundamental concepts regarding the regulation of gene expression and to examine a few real examples of those concepts in action.
Suggested discussion
Why is it important to regulate gene expression why not just express all genes all of the time?
Create a list of hypotheses with your classmates of reasons why the regulation of gene expression is important for bacteria and archaea and for eukaryotes. Instead of bacteria vs. eukaryotes, you may also want to consider contrasting reasons gene regulation is important for unicellular organisms versus multi-cellular organisms or communities of unicellular organisms (like colonies of bacteria).
Subproblems for transcription and the activity or RNA polymerase
Let us consider a protein coding gene and work through some logic. We start by imagining a simple case, where a protein-coding gene is encoded by a single contiguous stretch of DNA. We know that to transcribe this gene an RNA polymerase will need to be recruited to the start of the coding region. The RNA polymerase is not "smart" per se. There needs to be some mechanism, based on chemical logic, to help recruit the RNA polymerase to the start of the protein coding gene. Likewise, if this process is to be regulated, there needs to be some mechanism, or mechanisms, to dictate when an RNA polymerase should be recruited to the start of a gene, when it should not, and/or if it is recruited to the DNA whether or not it should actually begin transcription and how many times this process should happen. Note, that the previous sentence, has several distinct subproblems/questions (e.g. when is the polymerase recruited?; if recruited, should it start transcription?; if it starts transcription, how many times should this process repeat?). We can also reasonably infer, that there will need to be some mechanisms to "instruct" (more anthropomorphisms) the polymerase to stop transcription. Finally, since the role of transcription is to create RNA copies of the genome segments, we should also consider problems/questions related to other factors that influence the abundance of RNA, like mechanisms of degradation. There must be some mechanisms and these will likely be involved in the regulation of this process.
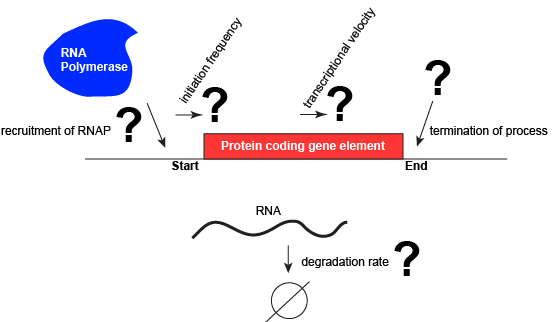
A schematic showing a protein coding gene and some of the questions or problems that we need to ask ourselves or alternatively problems we need to know solutions for if we are to understand how regulation of the transcriptional portion of the gene's expression is regulated.
Attribution: Marc T. Facciotti (own work)
Activation and Repression of Transcription
Some basics
Let us consider a protein coding gene and work through some logic. We start by imagining a simple case, where a protein-coding gene is encoded by a single contiguous stretch of DNA. We know that to transcribe this gene an RNA polymerase will need to be recruited to the start of the coding region. The RNA polymerase is not "smart" per se. There needs to be some mechanism, based on chemical logic, to help recruit the RNA polymerase to the start of the protein coding gene. Likewise, if this process is to be regulated, there needs to be some mechanism, or mechanisms, to dictate when an RNA polymerase should be recruited to the start of a gene, when it should not, and/or if it is recruited to the DNA, whether or not it should actually begin transcription and how many times this process should happen. Note, that the previous sentence, has several distinct subproblems/questions (e.g. when is the polymerase recruited?, if recruited should it start transcription?, if it starts transcription, how many times should this process repeat?). We can also reasonably infer, that there will need to be some mechanisms to "instruct" (more anthropomorphisms) the polymerase to stop transcription.
Recruiting RNA polymerase to specific sites
To initiate transcription, the RNA polymerase must be recruited to a segment of DNA near the start of a region of DNA encoding a functional transcript. The function of the RNA polymerase as described so far, however, is not to bind specific sequences but rather to move along any segment of DNA. Finding a way to recruit the polymerase to a specific site therefore seems contradictory to its usual behavior. Explaining this contradiction requires us to invoke something new. Either transcription can start anywhere and just those events that lead to a full productive transcript do anything useuful or something other than the RNA polymerase itself helps to recruit the enzyme to the beginning of a gene. The latter, we now take for granted, is indeed the case.
The recruitment of the RNA polymerase is mediated by proteins called general transcription factors. In bacteria, they have a special name: sigma factors. In archaea they are called TATA binding protein and transcription factor IIB. In eukaryotes, relatives of the archaeal proteins function in conjunction with numerous others to recruit the RNA polymerase. The general transcription factors have at least two basic functions: (1) They are able to chemically recognize a specific sequence of DNA and (2) they are able to bind the RNA polymerase. Together these two functions of general transcription factors solve the problem of recruiting an enzyme that is otherwise not capable of binding a specific DNA site. In some texts, the general transcription factors (and particularly the sigma factor varieties) are said to be part of the RNA polymerase. While they are certainly part of the complex when they help to target the RNA polymerase they do not continue with the RNA polymerase after it starts transcription.
The DNA site to which an RNA polymerase is recruited has a special name. It is called a promoter. While the DNA sequences of different promoters need not be exactly the same, different promoter sequences typically do have some special chemical properties in common. Obviously, one property is that they are able to associate with an RNA polymerase. In addition, the promoter usually has a DNA sequence that facilitates the dissociation of the double stranded DNA such that the polymerase can begin reading and transcription the coding region. (Note: technically we could have broken down the properties of the promoter into design challenge subproblems. In this case we skipped it, but you should still be able to step backwards and create the problem statements and or relevant questions once you find out about promoters).
In nearly all cases, but particularly in eukaryotic systems the complex of proteins that assembles with the RNA polymerase at promoters (typically called the pre-initiation complex) can number in the tens of proteins. Each of these other proteins has specific function but this is far to too much detail to dive into for Bis2A.
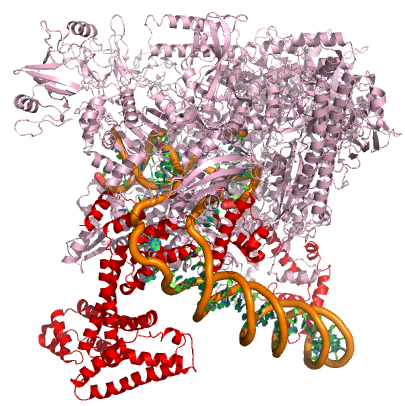
A model of the E. coli pre-initiation complex. The sigma factor is colored red. The DNA is depicted as orange tubes and opposing ring structures. The rest of the pre-initiation complex is colored pink. Note that the DNA has regions of double helix and an open structure inside the PIC.
Attribution: Structure derived from PDB coordinates (4YLN) Marc T. Facciotti (own work)
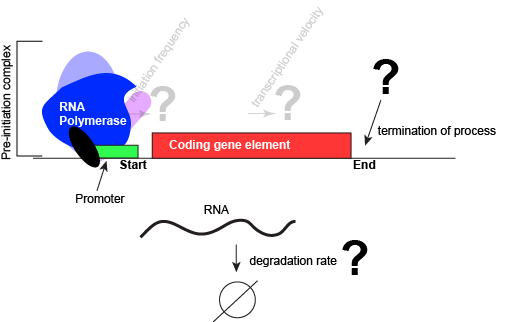
An abstract model of a generic transcriptional unit shown above with the addition of a promoter and PIC. Questions noted earlier that will likely not be covered in Bis2a have been grayed out.
Attribution: Marc T. Facciotti (own work)
Suggested discussion
How does general transcription factor recognize a specific sequence of DNA? What might be the chemical basis for this? How, by contrast might the interaction of a protein or enzyme that interacts with DNA non-specifically differ? Think about functional groups and propose a hypothesis.
States of a regulated promoter
Since promoters recruit an RNA polymerase these sites and the assembly of the pre-initiation complex are obvious sites for regulating the first steps of gene expression. At the level of transcription initiation, we often classify promoter into one of three classes. The first is called constitutive. Constitutive promoters are generally not regulated very strongly. Their base state is "on". When the constitutive transcription from a promoter is very high (relative to most other promoters), we will colloquially call that promoter a "strong constitutive" promoter. By contrast, if the amount of transcription from a constitutive promoter is low (relative to most other promoters) we will call that promoter a "weak constitutive" promoter.
A second way to classify promoters by the use of the term activated or equivalently, induced. These interchangeable terms are used to describe promoters that are sensitive to some external stimulus and respond to said stimulus by increasing transcription. Activated promoters have a base state generally exhibits little to no transcription. Transcription is then "activated" in response to a stimulus - the stimulus turns the promoter "on".
Finally, the third term used to classify promoters is by the use of the term repressed. These promoters also respond to stimuli but do so by decreasing transcription. The base state for these promoters is some basal level of transcription and the stimulus acts to turn down or repress transcription. Transcription is "repressed" in response to a stimulus - the stimulus turns the promoter "off".
The examples given above assumed that a single stimulus acts to regulate promoters. While this is the simplest case, many promoters may integrate different types of information and may be alternately activated by some stimuli and repressed by other stimuli.
Transcription factors help to regulate the behavior of a promoter
How are promoters sensitive to external stimuli? Mechanistically, in both activation and repression, require regulatory proteins to change the transcriptional output of the gene being observed. The proteins responsible for helping to regulate expression are generally called transcription factors. The specific DNA sequences bound by transcription factors are often called operators and in many cases the operators are very close to the promoter sequences.
Here's where the nomenclature gets potentially confusing - particularly when comparing across bacterial and eukaryotic research. In bacterial research, if the transcription factor acts by binding DNA and the RNA polymerase in a way that increases transcription, then it is typically called an activator. If, by contrast, the transcription factor acts by binding DNA to repress or decrease transcription of the gene then it is called a repressor.
Why are the classifications of activator and repressor potentially problematic? These terms, describe in idealized single functions. While this may be true in the case of some transcription factors, in reality other transcription factors may act to activate gene expression in some conditions while repressing in other conditions. Some transcription factors will simply act to modulate expression either up or down depending on context rather than shutting transcription "off" or turning it completely "on". To circumvent some of this possible confusion, some of your instructors prefer to avoid using the terms activator and repressor and instead prefer to simply discuss the activity of transcription various transcription factors as either a positive or a negative influence on gene expression in specific cases. If these terms are used, you might hear your instructor saying that the transcription factor in question ACTS LIKE/AS a repressor or that it ACTS LIKE/AS an activator, taking care not to call it simply an activator or repressor. It is more likely however that you will hear them say that a transcription factor is acting to positively or negatively influence transcription.
CAUTION: Depending on your instructor, you may cover a few real biological examples of positive and negative regulatory mechanisms. These specific examples will use the common names of the transcription factors - since the the examples will typically be drawn from the bacterial literature, the names of the transcription factors may include the terms "repressor" or "activator". These names are relics of when they were first discovered. Try to spend more time examining the logic of how the system works than trying to commit to memory any special properties of that specific protein to all other cases with the same label. That is, just because a protein is labeled as a repressor does not mean that it exclusively acts as a negative regulator in all cases or that it interacts with external signals in the exact same was as the example.
Suggested discussion
What types of interactions do you think happen between the amino acids of the transcription factor and the double helix of the DNA? How do transcription factors recognize their binding site on the DNA?
Allosteric Modulators of Regulatory Proteins
The activity of many proteins, including regulatory proteins and various transcription factors, can be allosterically modulated various factors, including by the relative abundance of small molecules in the cell. These small molecules are often referred to as inducers or co-repressorsor co-activators and are often metabolites, such as lactose or tryptophan or small regulatory molecules, such as cAMP or GTP. These interactions allow the TF to be responsive to environmental conditions and to modulate its function accordingly. It is helping to make a decision about whether to transcribe a gene or not depending on the abundance of the environmental signal.
Let us imagine a negative transcriptional regulator. In the most simple case we've considered so far, transcription of gene with a binding site for this transcription factor would be low when the TF is present and high when the TF is absent. We can now add a small molecule to this model. In this case the small molecule is able to bind the negative transcriptional regulator through sets of complementary hydrogen and ionic bonds. In this first example we will consider the case where the binding of the small molecule to the TF induces a conformational change to the TF that severely reduces its ability to bind DNA. If this is the case, the negative regulator - once bound by its small molecule - would release from the DNA. This would thereby relieve the negative influence and lead to increased transcription. This regulatory logic might be appropriate to have evolved in the following scenario: a small molecule food-stuff is typically absent from the environment. Therefore, genes encoding enzymes that will degrade/use that food should be kept "off" most of the time to preserve the cellular energy that their synthesis would use. This could be accomplished by the action of a negative regulator. When the food-stuff appears in the environment it would be appropriate for the enzymes responsible for its processing to be expressed. The food-stuff could then act by binding to the negative regulator, changing the TF's conformation, causing its release from the DNA and turning on transcription of the processing enzymes.
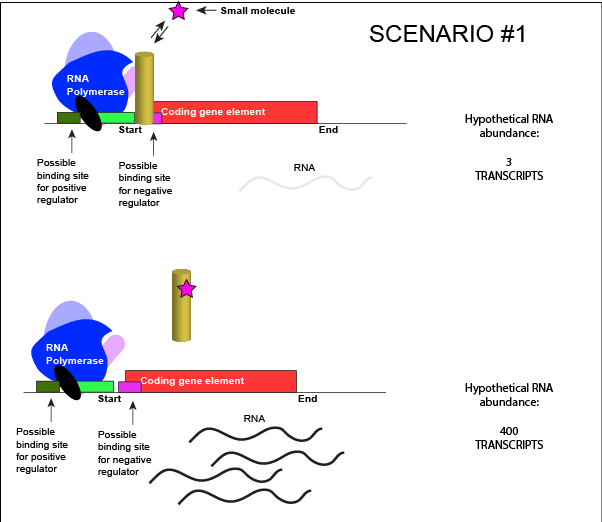
An abstract model of a generic transcriptional unit regulated by a negative regulator whose activity is modulated by a small molecule (depicted by a star). In this case, binding of the small molecule causes the TF to release from the DNA.
Attribution: Marc T. Facciotti (own work)
We can consider a second model for how a negatively acting TF might interact with a small molecule. In this case, the TF alone is unable to bind its regulatory site on the DNA. However, when a small molecule binds to the TF a conformational change occurs that reorients DNA binding amino-acids into the "correct" orientation for DNA binding. The TF-small molecule complex now binds to the DNA and acts to negatively influence transcription.
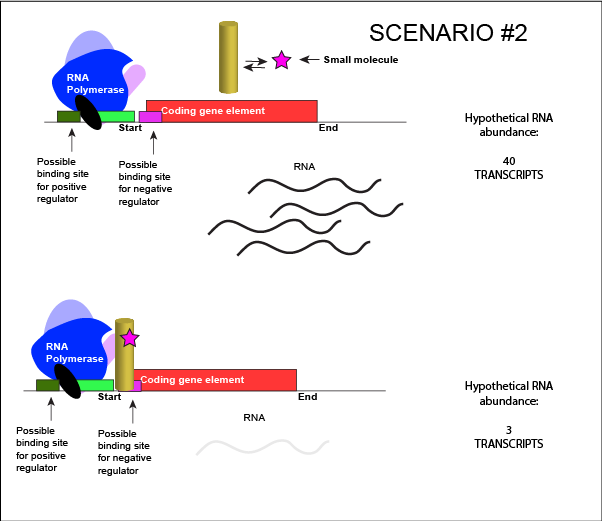
An abstract model of a generic transcriptional unit regulated by a negative regulator whose activity is modulated by a small molecule (depicted by a star). In this case, binding of the small molecule causes the TF to bind to the DNA.
Attribution: Marc T. Facciotti (own work)
Note how the activity of the TF can be modulated in distinctly different ways by a small molecule. Depending on the protein, the binding of this external signal can either cause binding of the TF-small molecule complex to DNA OR binding of the small molecule can cause the release of the TF-small molecule complex from the DNA. The same types of examples can be worked up for a positive regulator.
In both cases proposed above, the binding of a small molecule to a TF will be dependent on how strongly the TF interacts with the small molecule. This will depend on the types and spatial orientation of the protein's chemical functional groups, their protonation states (if applicable), and the complementary functional groups on the small molecule. It should not be surprising, therefore, to learn that the binding of the small molecule to the TF will be dependent on various factors, including but not limited to the relative concentrations of small-molecule and TF and pH.
Is it positive or negative regulation?
Resolving a common point of confusion
At this point, it is not uncommon for many Bis2a students to be slightly confused about how to determine if a transcription factor is acting as a positive or negative regulator. This confusion often comes after a discussion of the possible modes that stimulus (i.e. small molecule) can influence the activity of a transcription factor. This is not too surprising. In the examples above, the binding of a effector molecule to a transcription factor could have one of two different effects: (1) binding of the effector molecule could induce a DNA-bound transcription factor to release from its binding site, derepressing a promoter, and "turning on" gene expression. (2) binding of the effector molecule to the transcription factor could induce the TF to bind to its DNA binding site, repressing transcription and "turning off" gene expression. In the first case it might appear that the TF is acting to positively regulate expression, while in the second example it might appear that the TF is acting negatively.
However, in both examples above, the TF is acting as a negative regulator. To determine this we look at what happens when the TF binds DNA (whether a small molecule is bound to the TF or not). In both cases, binding of the TF to DNA represses transcription. The TF is therefore acting as a negative regulator. A similar analysis can be done with positively acting TFs.
Note that in some cases a TF may act as a positive regulator at one promoter and negative regulator at a different promoter so describing the behavior of the TF on a per case basis is often important (reading too much from the name it has been assigned can be misleading sometimes). Other TF protein can act alternately as both positive or negative regulators of the same promoter depending on conditions. Again, describing the behavior of the TF specifically for each case is advised.
A genetic test for positive or negative regulatory function of a TF
How does one determine if a regulatory protein functions in a positive or negative way? A simple genetic test is to ask "what happens to expression if the regulatory protein is absent?" This can be accomplished by removing the coding gene for the transcription factor from the genome. If a transcription factor acts positively, then its presence is required to activate transcription. In its absence, there is no regulatory protein, therefore no activation, and the outcome is lower transcription levels of a target gene. The opposite is true for a transcription factor acting negatively. In its absence expression should be increased, because the gene keeping expression low is no longer around.
Termination of Transcription and RNA degradation
Termination of transcription in bacteria
Once a gene is transcribed, the bacterial polymerase needs to be instructed to dissociate from the DNA template and liberate the newly made mRNA. Depending on the gene being transcribed, there are two kinds of termination signals. One is protein-based and the other is RNA-based. Rho-dependent termination is controlled by the rho protein, which tracks along behind the polymerase on the growing mRNA chain. Near the end of the gene, the polymerase encounters a run of G nucleotides on the DNA template and it stalls. As a result, the rho protein collides with the polymerase. The interaction with rho releases the mRNA from the transcription bubble.
Rho-independent termination is controlled by specific sequences in the DNA template strand. As the polymerase nears the end of the gene being transcribed, it encounters a region rich in C–G nucleotides. The mRNA folds back on itself, and the complementary C–G nucleotides bind together. The result is a stable hairpin that causes the polymerase to stall as soon as it begins to transcribe a region rich in A–T nucleotides. The complementary U–A region of the mRNA transcript forms only a weak interaction with the template DNA. This, coupled with the stalled polymerase, induces enough instability for the core enzyme to break away and liberate the new mRNA transcript.
Termination of transcription in eukaryotes
The termination of transcription is different for the different polymerases. Unlike in prokaryotes, elongation by RNA polymerase II in eukaryotes takes place 1,000–2,000 nucleotides beyond the end of the gene being transcribed. This pre-mRNA tail is subsequently removed by cleavage during mRNA processing. On the other hand, RNA polymerases I and III require termination signals. Genes transcribed by RNA polymerase I contain a specific 18-nucleotide sequence that is recognized by a termination protein. The process of termination in RNA polymerase III involves an mRNA hairpin similar to rho-independent termination of transcription in prokaryotes.
Termination of transcription in archaea
Termination of transcription in the archaea is far less studied than in the other two domains of life and is still not well understood. While the functional details are likely to resemble mechanisms that have been seen in the other domains of life the details are beyond the scope of this course.
Degradation of RNA
The lifetimes of different RNA species in the cell can vary dramatically, from seconds to hours. The mean lifetime of mRNA can also vary dramatically depending on the organism. For instance, the median lifetime for mRNA in E. coli is ~5 minutes. The half-life of mRNA in yeast is ~20 minutes and 600 minutes for human cells. Some of the degradation is "targeted". That is, some transcripts include a short sequence that targets them for RNA degrading enzymes, speeding the degradation rate. It doesn't take too much imagination to infer that this process might also be evolutionarily tuned for different genes. Simply realizing that degradation - and the tuning of degradation - can also be a factor in controlling the expression of a gene is sufficient for Bis2a.
Tuning termination
Like all other biological processes, the termination of transcription is not perfect. Sometimes, the RNA polymerase is able to read through terminator sequences and transcribe adjacent genes. This is particularly true in bacterial and archaeal genomes where the density of coding genes is high. This transcriptional read-through can have various effects but the most common two outcomes are: (1) If the adjacent genes are encoded on the same DNA strand as the actively transcribed gene, the adjacent genes may also be transcribed. (2) If the adjacent genes are transcribed on the opposite strand of the actively transcribed genes, RNA polymerase read-through by interfere with polymerases actively transcribing the neighboring gene. Not surprisingly, biology has in some instances evolved mechanisms that both act to minimize the influence of read-through and to take advantage of it. Therefore, the "strength" of a terminator - and its effectiveness of terminating transcription in a particular direction - are "tuned" by Nature and "used" (note the anthropomorphism in quotes) to regulate the expression of genes.
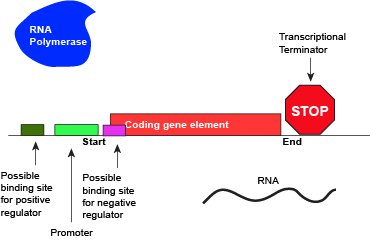
An abstract model of a full basic transcriptional unit and the various "parts" encoded on the DNA that may influence the expression of the gene. We expect students in Bis2A to be able to recreate a similar conceptual figure from memory.
Attribution: Marc T. Facciotti (own work)
Summary of gene regulation
In the preceding text we have examined several ways to start solving some of the design challenges associated with regulating the amount of transcript that is created for a single coding region of the genome. We have looked in abstract terms at some of the processes responsible for controlling the initiation of transcription, how these may be made sensitive to environmental factors, and very briefly at the processes that terminate transcription and handle the active degradation of RNA. We have avoided more Each of these processes can be quantitatively tuned by nature to be "stronger" or "weaker". It is important to realize that the real values of "strength" (e.g. promoter strength, degradation rates, etc.) influence the behavior of the overall process in potentially functionally important ways.
Examples of Bacterial Gene Regulation
This section describes two examples of transcriptional regulation in bacteria. These are presented as illustrative examples. Use these examples to learn some basic principles about mechanisms of transcriptional regulation. Be on the lookout in class, in discussion, and in the study-guides for extensions of these ideas and use these to explain the regulatory mechanisms used for regulating other genes.
Gene Regulation Examples in E. coli
The DNA of bacteria and archaea are usually organized into one or more circular chromosomes in the cytoplasm. The dense aggregate of DNA that can be seen in electron micrographs is called the nucleoid. In bacteria and archaea, genes, whose expression needs to be tightly coordinated (e.g. genes encoding proteins that are involved in the same biochemical pathway) are often grouped closely together in the genome. When the expression of multiple genes is controlled by the same promoter and a single transcript is produced these expression units are called operons. For example, in the bacterium Escherschia coli all of the genes needed to utilize lactose are encoded next to one another in the genome. This arrangement is called the lactose (or lac) operon. It is often the case in bacteria and archaea that nearly 50% of all genes are encoded into operons of two or more genes.
The Role of the Promoter
The first level of control of gene expression is at the promoter itself. Some promoters recruit RNA polymerase and turn those DNA-protein binding events into transcripts more efficiently than other promoters. This intrinsic property of a promoter, it's ability to produce transcript at a particular rate, is referred to as promoter strength. The stronger the promoter, the more RNA is made in any given time period. Promoter strength can be "tuned" by Nature in very small or very large steps by changing the nucleotide sequence the promoter (e.g. mutating the promoter). This results in families of promoters with different strengths that can be used to control the maximum rate of gene expression for certain genes.
UC Davis Undergraduate Connection:
A group of UC Davis students interested in synthetic biology used this idea to create synthetic promoter libraries for engineering microbes as part of their design project for the 2011 iGEM competition.
Example #1: Trp Operon
Logic for regulating tryptophan biosynthesis
E. coli, like all organisms, needs to either synthesize or consume amino acids to survive. The amino acid tryptophan is one such amino acid. E. colican either import tryptophan from the environment (eating what it can scavenge from the world around it) or synthesize tryptophan de novo using enzymes that are encoded by five genes. These five genes are encoded next to each other in the E. coli genome into what is called the tryptophan (trp) operon (Figure below). If tryptophan is present in the environment, then E. coli does not need to synthesize it and the switch controlling the activation of the genes in the trp operon is switched off. However, when environmental tryptophan availability is low, the switch controlling the operon is turned on, transcription is initiated, the genes are expressed, and tryptophan is synthesized. See the figure and paragraphs below for a mechanistic explanation.
Organization of the trp operon
Five genomic regions encoding tryptophan biosynthesis enzymes are arranged sequentially on the chromosome and are under the control of a single promoter - they are organized into an operon. Just before the coding region is the transcriptional start site. This is, as the name implies, the location where the RNA polymerase starts a new transcript. The promoter sequence is further upstream of the transcriptional start site.
A DNA sequence called an "operator" is also encoded between the promoter and the first trp coding gene. This operator is the DNA sequence to which the transcription factor protein will bind.
A few more details regarding TF binding sites
It should be noted that the use of the term "operator" is limited to just a few regulatory systems and almost always refers to the binding site for a negatively acting transcription factor. Conceptually what you need to remember is that there are sites on the DNA that interact with regulatory proteins allowing them to perform their appropriate function (e.g. repress or activate transcription). This theme will be repeated universally across biology whether the "operator" term is used or not.
Moreover, while the specific examples you will be show depict TF binding sites in their known locations, these locations are not universal to all systems. Transcription factor binding sites can vary in location relative to the promoter. There are some patterns (e.g. positive regulators are often upstream of the promoter and negative regulators bind downstream), but these generalizations are not true for all cases. Again, the key thing to remember is that transcription factors (both positive and negatively acting) have binding sites with which they interact to help regulate the initiation of transcription by RNA polymerase.
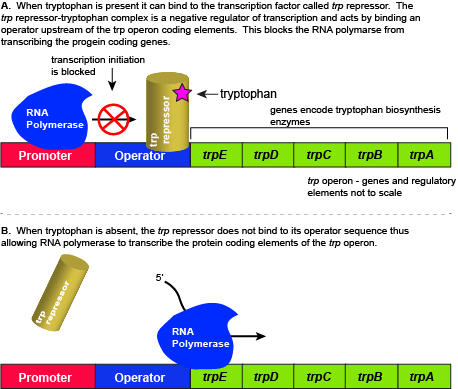
The five genes that are needed to synthesize tryptophan in E. coli are located next to each other in the trp operon. When tryptophan is plentiful, two tryptophan molecules bind to the transcription factor and allow the TF-tryptophan complex to bind at the operator sequence. This physically blocks the RNA polymerase from transcribing the tryptophan biosynthesis genes. When tryptophan is absent, the transcription factor does not bind to the operator and the genes are transcribed.
Attribution: Marc T. Facciotti (own work)
Regulation of the trp operon
When tryptophan is present in the cell: two tryptophan molecules bind to the trp repressor protein. When tryptophan binds to the transcription factor it causes a conformational change in the protein which now allows the TF-tryptophan complex to bind to the trp operator sequence. Binding of the tryptophan–repressor complex at the operator physically prevents the RNA polymerase from binding, and transcribing the downstream genes. When tryptophan is not present in the cell, the transcription factor does not bind to the operator; therefore, the transcription proceeds, the tryptophan utilization genes are transcribed and translated, and tryptophan is thus synthesized.
Since the transcription factor actively binds to the operator to keep the genes turned off, the trp operon is said to be "negatively regulated" and the proteins that bind to the operator to silence trp expression are negative regulators.
Suggested discussion
Do you think that the constitutive expression levels of the trp operon are high or low? Why?
Suggestion discussion
Suppose nature took a different approach to regulating the trp operon. Design a method for regulating the expression of the trp operon with a positive regulator instead of a negative regulator. (hint: we ask this kind of question all of the time on exams)
External link
Watch this video to learn more about the trp operon.
Example #2: The lac operon
Rationale for studying the lac operon
In this example, we examine the regulation of genes encoding proteins whose physiological role is to import and assimilate the disaccharide lactose, the lac operon. The story of the regulation of lac operon is a common example used in many introductory biology classes to illustrate basic principles of inducible gene regulation. We choose to describe this example second because it is, in our estimation, more complicated than the previous example involving the activity of a single negatively acting transcription factor. By contrast, the regulation of the lac operon is, in our opinion, a wonderful example of how the coordinated activity of both positive and negative regulators around the same promoter can be used to integrate multiple different sources of cellular information to regulate the expression of genes.
As you go through this example, keep in mind the last point. For many Bis2a instructors it is more important for you to learn the lac operon story than it is to know the logic table presented below. For those instructors for which this is the case, they will usually make a point to let you know and often deliberately not include exam questions about the lac operon. Rather they will test you on whether you understood the basis underlying the regulatory mechanisms that you study. If it's not clear what the instructor wants you should ask.
The utilization of lactose
Lactose is a disaccharide composed of the hexoses glucose and galactose. It is commonly found in high abundance in milk and some milk products. As one can imagine, the disaccharide can be an important food-stuff for microbes that are able to utilize its two hexoses. E. coli is able to use multiple different sugars as energy and carbon sources, including lactose and the lac operon is a structure that encodes the genes necessary to acquire and process lactose from the local environment. Lactose, however, has not been frequently encountered by E. coli during its evolution and therefore the genes of the lac operon must typically be repressed (i.e. "turned off") when lactose is absent. Driving transcription of these genes when lactose is absent would waste precious cellular energy. By contrast, when lactose is present, it would make logical sense for the genes responsible for the utilization of the sugar to be expressed (i.e. "turned on"). So far the story is very similar to that of the tryptophan operon described above.
However, there is a catch. Experiments conducted in the 1950's by Jacob and Monod clearly demonstrated that E. coli prefers to utilize all the glucose present in the environment before it begins to utilize lactose. This means that the mechanism used to decide whether or not to express the lactose utilization genes must be able to integrate two types of information (1) the concentration of glucose and (2) the concentration of lactose. While this could theoretically be accomplished in multiple ways, we will examine how the lac operon accomplishes this by using multiple transcription factors.
The transcriptional regulators of the lac operon
The lac repressor - a direct sensor of lactose
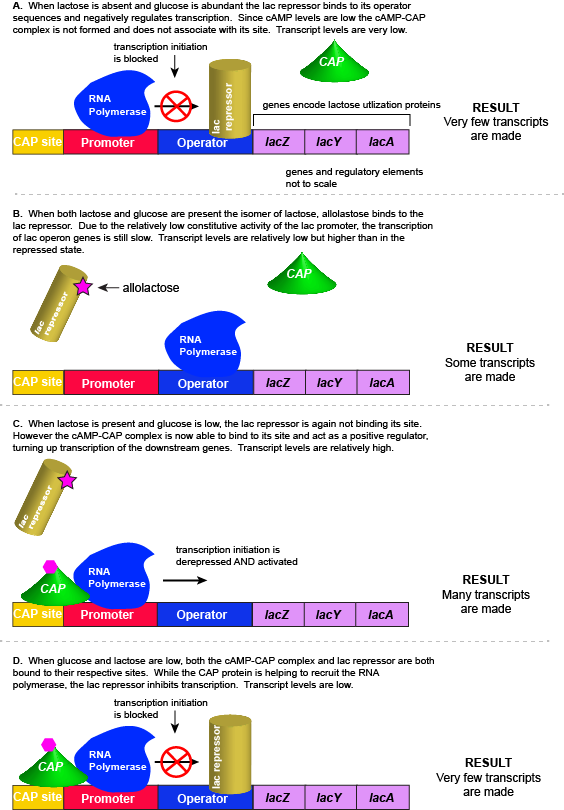
As noted, the lac operon normally has very low to no transcriptional output in the absence of lactose. This is due to two factors: (1) the constitutive promoter strength for the operon is relatively low and (2) the constant presence of the LacI repressor protein negatively influences transcription. This protein binds to the operator site near the promoter and blocks RNA polymerase from transcribing the lac operon genes. By contrast, if lactose is present, lactose will bind to the LacI protein, inducing a conformational change that prevents LacI-lactose complex from binding to its binding sites. Therefore, when lactose is present the negative regulatory LacI is not bound to the its binding site and transcription of lactose utilizing genes can proceed.
CAP protein - an indirect sensor of glucose
In E. coli, when glucose levels drop, the small molecule cyclic AMP (cAMP) begins to accumulate in the cell. cAMP is a common signaling molecule that is involved in glucose and energy metabolism in many organisms. When glucose levels decline in the cell, the increasing concentrations of cAMP allow this compound to bind to the positive transcriptional regulator called catabolite activator protein (CAP) - also referred to as CRP. cAMP-CAP complex has many sites located throughout the E. coli genome and many of these sites are located near the promoters of many operons that control the processing of various sugars.
In the lac operon, the cAMP-CAP binding site is located upstream of the promoter. Binding of cAMP-CAP to the DNA helps to recruit and retain RNA polymerase to the promoter. The increased occupancy of RNA polymerase to its promoter, in turn, results in increased transcriptional output. In this case the CAP protein is acting as a positive regulator.
Note that the CAP-cAMP complex can, in other operons, also act as a negative regulator depending upon where the binding site for CAP-cAMP complex is located relative to the RNA polymerase binding site.
Putting it all together: Inducing expression of the lac operon
For the lac operon to be activated, two conditions must be met. First, the level of glucose must be very low or non-existent. Second, lactose must be present. Only when glucose is absent and lactose is present will the lac operon be transcribed. When this condition is achieved the LacI-lactose complex dissociates the negative regulator from near the promoter, freeing the RNA polymerase to transcribe the operon's genes. Moreover, high cAMP (indirectly indicative of low glucose) levels trigger the formation of the CAP-cAMP complex. This TF-inducer pair now bind near the promoter and act to positively recruit the RNA polymerase. This added positive influence boosts transcriptional output and lactose can be efficiently utilized. The mechanistic output of other combinations of binary glucose and lactose conditions are descried in the table below and in the figure that follows.
A more nuanced view of lac repressor function
The description of the lac repressor's function correctly describes the logic of the control mechanism used around the lac promoter. However, the molecular description of binding sites is a bit overly simplified. In reality the lac repressor has three similar, but not identical, binding sites called Operator 1, Operator 2, and Operator 3. Operator 1 is very close to the transcript start site (denoted +1). Operator 2 is located about +400nt into the coding region of the LacZ protein. Operator 3 is located about -80nt before the transcript start site (just "outside" of the CAP binding site).
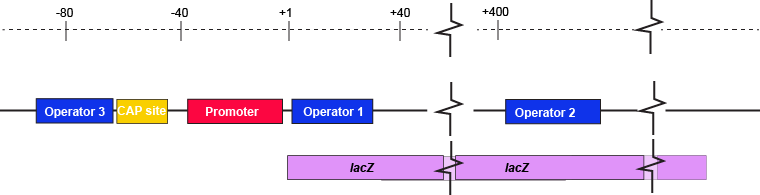
The lac operon regulatory region depicting the promoter, three lac operators, and CAP binding site. The coding region for the Lac Z protein is also shown relative to the operator sequences. Note that two of the operators are in the protein coding region - there are multiple different types of information simultaneously encoded in the DNA.
Attribution: Marc T. Facciotti (own work)
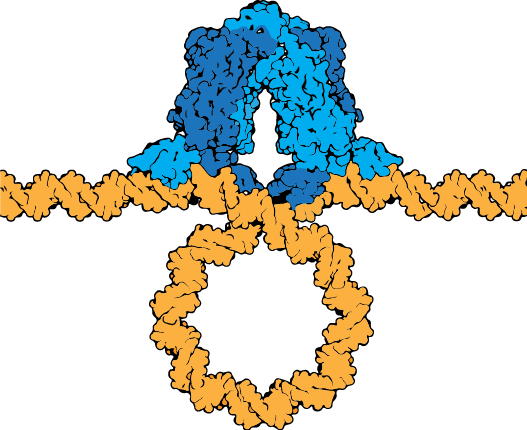
The lac repressor tetramer (blue) depicted binding two operators on a strand of looped DNA (orange).
Attribution: Marc T. Facciotti (own work) - Adapted from Goodsell (https://pdb101.rcsb.org/motm/39)
Eukaryotic Gene Regulation
Regulation overview
As was previously noted, regulation is all about decision making. Gene regulation, as a general topic, is related to making decisions about the functional expression of genetic material. Whether the final product is an RNA species or a protein, the production of the final expressed product requires processes that take multiple steps. We have spent some time discussing some of these steps (i.e. transcription and translation) and some of the mechanisms that nature uses for sensing cellular and environmental information to regulate the initiation of transcription.
When we discussed the concept of strong and weak promoters we introduced the idea that regulating the amount (number of molecules) of transcript that was produced from a promoter in some unit of time might also be important for function. This should not be entirely surprising. For a protein coding gene, the more transcript that is produced, the greater potential there is to make more protein. This might be important in cases where making a lot of a particular enzyme is key for survival. By contrast, in other cases only a little protein is required and making too much would be a waste of cellular resources. In this case low levels of transcription might be preferred. Promoters of differing strengths can accommodate these varying needs. With regards to transcript number, we also briefly mentioned that synthesis is not the only way to regulate abundance. Degradation processes are also important to consider.
In this section, we add to these themes by focusing on eukaryotic regulatory processes. Specifically, we examine - and sometimes re-examine - some of the multiple steps that are required to express genetic material in eukaryotic organisms in the context of regulation. We want you not only to think about the processes but also to recognize that each step in the process of expression is also an opportunity to fine tune not only the abundance of a transcript or protein but also its functional state, form (or variant), and/or stability. Each of these additional factors may also be vitally important to consider for influencing the abundance of conditionally-specific functional variants.
Structural differences between bacterial and eukaryotic cells influencing gene regulation
The defining hallmark of the eukaryotic cell is the nucleus, a double membrane that encloses the cell's hereditary material. In order to efficiently fits the organism's DNA into the confined space of the nucleus, the DNA is first packaged and organized by protein into a structure called chromatin. This packaging of the nuclear material reduces access to specific parts of the chromatin. Indeed, some elements of the DNA are so tightly packed that the transcriptional machinery cannot access regulatory sites like promoters. This means that one of the first sites of transcriptional regulation in eukaryotes must be the control access to the DNA itself. Chromatin proteins can be subject to enzymatic modification that can influence whether they bind tightly (limited transcriptional access) or more loosely (greater transcriptional access) to a segment of DNA . This process of modification - whichever direction is considered first - is reversible. Therefore DNA can be dynamically sequestered and made available when the "time is right".
The regulation of gene expression in eukaryotes also involves some of the same additional fundamental mechanisms discussed in the module on bacterial regulation (i.e. the use of strong or weak promoters, transcription factors, terminators etc.) but the actual number of proteins involved is typically much greater in eukaryotes than bacteria or archaea.
The post-transcriptional enzymatic processing of RNA that occurs in the nucleus and the export of the mature mRNA to the cytosol are two additional difference between bacterial and eukaryotic gene regulation. We will consider this level of regulation in more detail below.
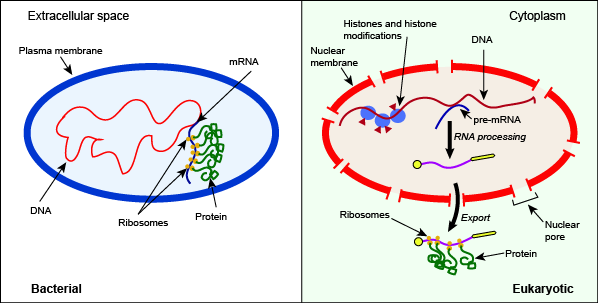
Depiction of some key differences between the processes of bacterial and eukaryotic gene expression. Note in this case the presence of histone and histone modifiers, the splicing of pre-mRNA, and the export of the mature RNA from the nucleus as key differentiators between the bacterial and eukaryotic systems.
Attribution: Marc T. Facciotti (own work)
DNA Packing and Epigenetic Markers
The DNA in eukaryotic cells is precisely wound, folded, and compacted into chromosomes so that it will fit into the nucleus. It is also organized so that specific segments of the chromosomes can be easily accessed as needed by the cell. Areas of the chromosomes that are more tightly compacted will be harder for proteins to bind and therefore lead to reduced gene expression of genes encoded in those areas. Regions of the genome that are loosely compacted will be easier for proteins to access, thus increasing the likelihood that the gene will be transcribed. Discussed here are the ways in which cells regulate the density of DNA compaction.
DNA packing
The first level of organization, or packing, is the winding of DNA strands around histone proteins. Histones package and order DNA into structural units called nucleosomes, which can control the access of proteins to specific DNA regions. Under the electron microscope, this winding of DNA around histone proteins to form nucleosomes looks like small beads on a string. These beads (nucleosome complexes) can move along the string (DNA) to alter which areas of the DNA are accessible to transcriptional machinery. While nucleosomes can move to open the chromosome structure to expose a segment of DNA, they do so in a very controlled manner.
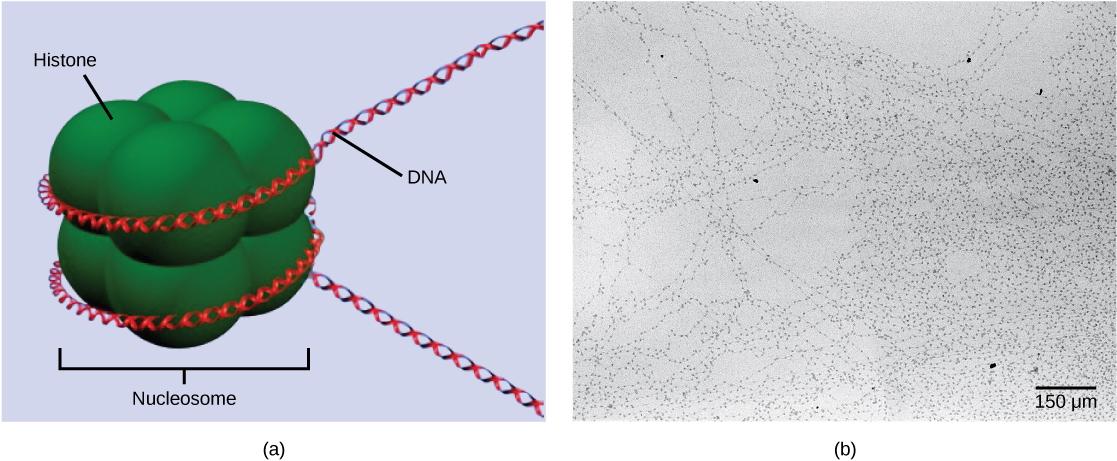
DNA is folded around histone proteins to create (a) nucleosome complexes. These nucleosomes control the access of proteins to the underlying DNA. When viewed through an electron microscope (b), the nucleosomes look like beads on a string. (credit “micrograph”: modification of work by Chris Woodcock)
Histone Modification
How the histone proteins move is dependent on chemical signals found on both the histone proteins and on the DNA. These chemical signals are chemical tags added to histone proteins and the DNA that tell the histones if a chromosomal region should be "open" or "closed". The figure below depicts modifications to histone proteins and DNA. These tags are not permanent, but may be added or removed as needed. They are chemical modifications (phosphate, methyl, or acetyl groups) that are attached to specific amino acids in the histone proteins or to the nucleotides of the DNA. The tags do not alter the DNA base sequence, but they do alter how tightly wound the DNA is around the histone proteins. DNA is a negatively charged molecule; therefore, changes in the charge of the histone will change how tightly wound the DNA molecule will be. When unmodified, the histone proteins have a large positive charge; by adding chemical modifications like acetyl groups, the charge becomes less positive.
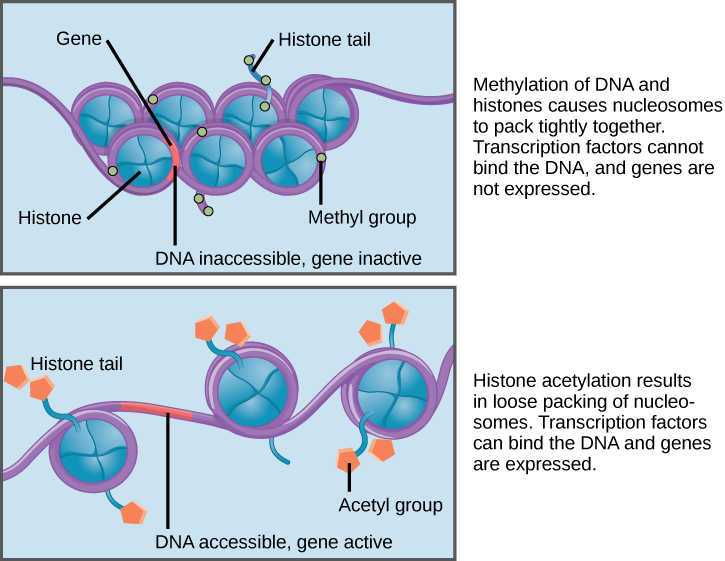
Nucleosomes can slide along DNA. When nucleosomes are spaced closely together (top), transcription factors cannot bind and gene expression is turned off. When the nucleosomes are spaced far apart (bottom), the DNA is exposed. Transcription factors can bind, allowing gene expression to occur. Modifications to the histones and DNA affect nucleosome spacing.
Suggested discussion
Why do histone proteins normally have a large amount of positive charges (histones contain a high number of lysine amino acids). Would removal of the positive charges cause a tightening of loosening of the histone-DNA interaction?
Suggested discussion
Predict the state of the histones in areas of the genome that are transcribed regularly. How do these differ from areas that do not experience high levels of transcription?
DNA Modification
The DNA molecule itself can also be modified. This occurs within very specific regions called CpG islands. These are stretches with a high frequency of cytosine and guanine dinucleotide DNA pairs (CG) often found in the promoter regions of genes. When this configuration exists, the cytosine member of the pair can be methylated (a methyl group is added). This modification changes how the DNA interacts with proteins, including the histone proteins that control access to the region. Highly methylated (hypermethylated) DNA regions with deacetylated histones are tightly coiled and transcriptionally inactive.
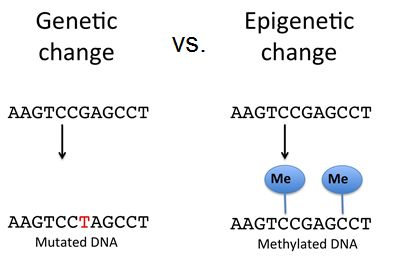
Epigenetic changes do not result in permanent changes in the DNA sequence. Epigenetic changes alter the chromatin structure (protein-DNA complex) to allow or deny access to transcribe genes. DNA modification such as methylation on cytosine nucleotides can either recruit repressor proteins that block RNA polymerase's access to transcribe a gene or they can aid in compacting the DNA to block all protein access to that area of the genome. These changes are reversible whereas mutations are not, however, epigenetic changes to the chromosome can also be inherited.
Source: modified from https://researcherblogski.wordpress....r/dudiwarsito/
Regulation of gene expression through chromatin remodeling is called epigenetic regulation. Epigenetic means “around genetics.” The changes that occur to the histone proteins and DNA do not alter the nucleotide sequence and are not permanent. Instead, these changes are temporary (although they often persist through multiple rounds of cell division and can be inherited) and alter the chromosomal structure (open or closed) as needed.
External link
View this video that describes how epigenetic regulation controls gene expression.
Eukaryotic gene structure and RNA processing
Eukaryotic gene structure
Many eukaryotic genes, particularly those encoding protein products, are encoded on the genome discontinuously. That is, the coding region is broken into pieces by intervening non-coding gene elements. The coding regions are termed exons while the intervening non-coding elements are termed introns. The figure below depicts a generic eukaryotic gene.
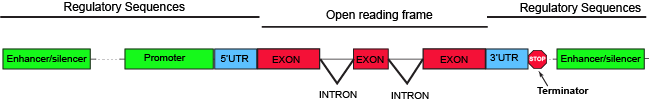
The parts of a typical discontinuous eukaryotic gene.
Attribution: Marc T. Facciotti (own work)
Parts of a generic eukaryotic gene include familiar elements like a promoter and terminator. Between those two elements, the region encoding all of the elements of the gene that have the potential to be translated (they have no stop codons), like in bacterial systems, is called the open reading frame (ORF). Enhancer and/or silencer elements are regions of the DNA that serve to recruit regulatory proteins. These can be relatively close to the promoter, like in bacterial systems, or thousands of nucleotides away. Also present in many bacterial transcripts, 5' and 3' untranslated regions (UTRs) also exist. These regions of the gene encode segments of the transcript, which, as their names imply, are not translated and sit 5' and 3', respectively, to the ORF. The UTRs typically encode some regulatory elements critical for regulating transcription or steps of gene expression that occur post-transcriptionally.
The RNA species resulting from the transcription of these genes are also discontinuous and must therefore be processed before exiting the nucleus to be translated or used in the cytosol as mature RNAs. In eukaryotic systems this includes RNA splicing, 5' capping, 3' end cleavage and polyadenylation. This series of steps is a complex molecular process that must occur within the closed confines of the nucleus. Each one of these steps provides an opportunity for regulating the abundance of exported transcripts and the functional forms that these transcripts will take. While these would be topics for more advanced courses, think about how to frame some of the following topics as subproblems of the Design Challenge of genetic regulation. If nothing else, begin to appreciate the highly orchestrated molecular dance that must occur to express a gene and how this is a stunning bit of evolutionary engineering.
5' capping
Like in bacterial systems, eukaryotic systems must assemble a pre-initiation complex at and around the promoter sequence to initiate transcription. The complexes that assemble in eukaryotes serve many of the same function as those in bacterial systems but they are significantly more complex, involving many more regulatory proteins. This added complexity allows for a greater degree of regulation and for the assembly of proteins with functions that occur predominantly in eukaryotic systems. One of these additional functions is the "capping" of nascent transcripts.
In eukaryotic protein coding genes, the RNA that is first produced is called the pre-mRNA. The "pre" prefix signifies that this is not the full mature mRNA that will be translated and that it first requires some processing. The modification known as 5'-capping occurs after the pre-mRNA is about 20-30 nucleotides in length. At this point the pre-RNA typically receives its first post-transcriptional modification, a 5'-cap. The "cap" is a chemical modification - a 7-methylguanosine - whose addition to the 5' end of the transcript is enzymatically catalyzed by multiple enzymes called the capping enzyme complex (CEC) a group of multiple enzymes that carry out sequential steps involved in adding the 5'-cap. The CEC binds to the RNA polymerase very early in transcription and carries out a modification of the 5' triphosphate, the subsequent transfer of at GTP to this end (connecting the two nucleotides using a unique 5'-to-5' linkage), the methylation of the newly transferred guanine, and in some transcripts the additional modifications to the first few nucleotides. This 5'-cap appears to function by protecting the emerging transcript from degradation and is quickly bound by RNA binding proteins known as the cap-binding complex (CBC). There is some evidence that this modification and the proteins bound to it play a role in targeting the transcript for export from the nucleus. Protecting the nascent RNA from degradation is not only important for conserving the energy invested in creating the transcript but is clearly involved in regulating the abundance of fully-functional transcript that is produced. Moreover, the role of the 5'-cap in guiding the transcript for export will directly help to regulate not only the amount of transcript that is made but, perhaps more importantly, the amount of transcript that is exported to the cytoplasm that has the potential to be translated.
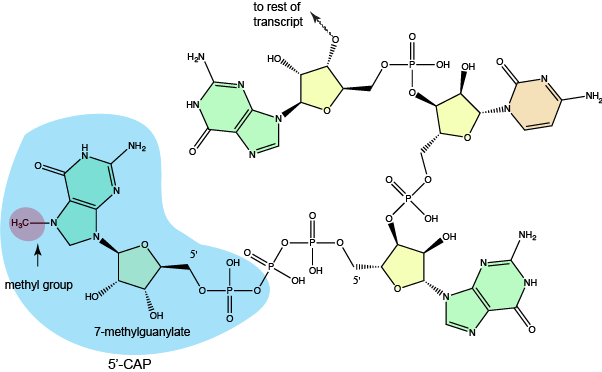
The structure of a typical 7-methylguanylate cap.
Attribution: Marc T. Facciotti (own work)
Transcript splicing
Nascent transcripts must be processed into mature RNAs by joining exons and removing the intervening introns. This is accomplished by a multicomponent complex of RNA and proteins called the spliceosome. The spliceosome complex assembles on the nascent transcript and in many cases the decisions about which introns to combine into a mature transcript are made at this point. How these decisions are made is still not completely understood but involves the recognition of specific DNA sequences at the splice sites by RNA and protein species and several catalytic events. It is interesting to note that the catalytic portion of the spliceosome is made of RNA rather than protein. Recall that the ribosome is another example of a RNA-protein complex where the RNA serves as the primary catalytic component. The selection of which splice variant to make is a form of regulating gene expression. In this case rather than simply influencing abundance of a transcript, alternative splicing allows the cell to make decisions about which form of transcript is made.
The alternative splice forms of genes that result in protein products of related structure but of varying function are known as isoforms. The creation of isoforms is common in eukaryotic systems and is known to be important in different stages of development in multicellular organisms and in defining the functions of different cell types. By encoding multiple possible gene products from a single gene whose transcription initiation is encoded from a single transcriptional regulatory site (by making the decision of which end-product to produce post-transcriptionally) obviates the need to create and maintain independent copies of each gene in different parts of the genome and evolving independent regulatory sites. Therefore, the ability to form multiple isoforms from a single coding region is though to be evolutionarily advantageous because it enables some efficiency in DNA coding, minimizes transcriptional regulatory complexity, and may lower the energy burden of maintaining more DNA and protecting it from mutation. Some examples of possible outcomes of alternative splicing can include: the generation of enzyme variants with differential substrate affinity or catalytic rates; signal sequences that target proteins to various sub-cellular compartments can be changed; entirely new functions, via the swapping of protein domains can be created. These are just a few examples.
One additional interesting possible outcome of alternative splicing is the introduction of stop codons that can, through a mechanism that seems to require translation, lead to the targeted decay of the transcript. This means that, in addition to the control of transcription initiation and 5'-capping, alternative splicing can also be considered one of the regulatory mechanisms that may influence transcript abundance. The effects of alternative splicing are therefore potentially broad - from complete loss of function to novel and diversified function to regulatory effects.
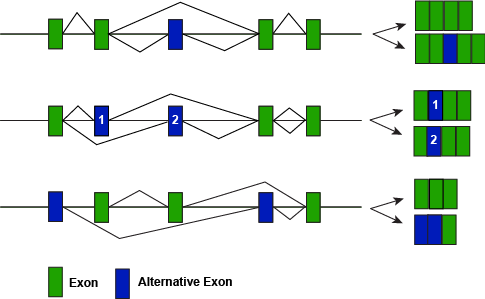
A figure depicting some of the different modes of alternative splicing illustrating how different splice variants can lead to different protein forms.
Attribution: Marc T. Facciotti (own work)
3' end cleavage and polyadenylation
One final modification is made to nascent pre-mRNAs before they leave the nucleus - the cleavage of the 3' end and its polyadenylation. This two step process is catalyzed by two different enzymes (as depicted below) and may decorate the 3' end of transcripts with up to nearly 200 nucleotides. This modification enhances the stability of the transcript. Generally, the more As in the polyA tag the longer lifetime that transcript has. The polyA tag also seems to play a role in the export of the transcript from the nucleus. Therefore, the 3' polyA tag plays a role in gene expression by regulating functional transcript abundance and how much is exported from the nucleus for translation.
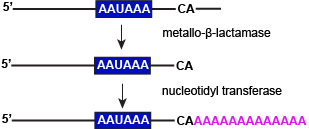
A two step process is involved in modifying the 3' ends of transcripts prior to nuclear exports. These include cutting transcripts just downstream of a conserved sequence (AAUAAA) and transferring adenylate groups. Both processes are enzymatically catalyzed.
Attribution: Marc T. Facciotti (own work)
microRNAs
RNA Stability and microRNAs
In addition to the modifications of the pre-RNA described above and the associated proteins that bind to the nascent and transcripts, there are other factors that can influence the stability of the RNA in the cell. One example are elements called microRNAs. The microRNAs, or miRNAs, are short RNA molecules that are only 21–24 nucleotides in length. The miRNAs are transcribed in the nucleus as longer pre-miRNAs. These pre-miRNAs are subsequently chopped into mature miRNAs by a protein called dicer. These mature miRNAs recognize a specific sequence of a target RNA through complementary base pairing. miRNAs, however, also associate with a ribonucleoprotein complex called the RNA-induced silencing complex (RISC). RISC binds a target mRNA, along with the miRNA, to degrade the target mRNA. Together, miRNAs and the RISC complex rapidly destroy the RNA molecule. As one might expect, the transcription of pre-miRNAs and their subsequent processing is also tightly regulated.
Nuclear export
Nuclear export
Fully processed, mature transcripts, must be exported through the nucleus. Not surprisingly this process involves the coordination of a mature RNA species to which are bound many accessory proteins - some of which have been intimately involved in the modifications discussed above - and a protein complex called the nuclear pore complex (NPC). Transport through the NPC allows flow of proteins and RNA species to move in both directions and is mediated by a number of proteins. This process can be used to selectively regulate the transport of various transcripts depending on which proteins associate with the transcript in question. This means that not all transcripts are treated equally by the NPC - depending on modification state and the proteins that have associated with a specific species of RNA it can be moved either more or less efficiently across the nuclear membrane. Since the rate of movement across the pore will influence the abundance of mature transcript that is exported into the cytosol for translation export control is another example of a step in the process of gene regulation that can be modulated. In addition, recent research has implicated interactions between the NPC and transcription factors in the regulation of transcription initiation, likely through some mechanism whereby the transcription factors tether themselves to the nuclear pores. This last example demonstrates how interconnected the regulation of gene expression is across the multiple steps of this complex process.
Many additional details of the processes described above are known to some level of detail, but many more questions remain to be answered. For the sake of Bis2a it is sufficient to begin forming a model of the steps that occur in the production of a mature transcript in eukaryotic organisms. We have painted a picture with very broad strokes, trying to present a scene that reflect what happens generally in all eukaryotes. In addition to learning the key differentiating features of eukaryotic gene regulation, we would also like for Bis2a students to begin thinking of each of these steps as an opportunity for Nature to regulate gene expression in some way and to be able to rationalize how deficiencies or changes in these pathways - potentially introduced through mutation - might influence gene expression.
While we did not explicitly bring up the Design Challenge or Energy Story here these formalisms are equally adept at helping you to make some sense of what is being described. We encourage you to try making an Energy Story for various processes. We also encourage you to use the Design Challenge rubric to reexamine the stories above: identify problems that need solving; hypothesize potential solutions and criteria for success. Use there formalisms to dig deeper and ask new questions/identify new problems or things that you don't know about the processes is what experts do. Chances are that doing this suggested exercise will lead you to identify a direction of research that someone has already pursued (you'll feel pretty smart about that!). Alternatively, you may raise some brand new question that no one has thought of yet.
Control of Protein Abundance
After an mRNA has been transported to the cytoplasm, it is translated into protein. Control of this process is largely dependent on the RNA molecule. As previously discussed, the stability of the RNA will have a large impact on its translation into a protein. As the stability changes, the amount of time that it is available for translation also changes.
The initiation complex and translation rate
Like transcription, translation is controlled by proteins complexes of proteins and nucleic acids that must associate to initiate the process. In translation, one of the first complexes that must assembles to start the process is referred to as the initiation complex. The first protein to bind to the mRNA that helps initiate translation is called eukaryotic initiation factor-2 (eIF-2). Activity of the eIF-2 protein is controlled by multiple factors. The first is whether or not it is bound to a molecule of GTP. When the eIF-2 is bound to GTP it is considered to be in an active form. The eIF-2 protein bound to GTP can bind to the small 40S ribosomal subunit. When bound, the eIF-2/40S ribosome complex, bringing with it the mRNA to be translated, also recruits the methionine initiator tRNA associates. At this point, when the initiator complex is assembled, the GTP is hydrolyzed into GDP creating an "inactive form of eIF-2 that is released, along with the inorganic phosphate, from the complex. This step, in turn, allows the large 60S ribosomal subunit to bind and to begin translating the RNA. The binding of eIF-2 to the RNA further controlled by protein phosphorylation. When eIF-2 is phosphorylated, it undergoes a conformational change and cannot bind to GTP thus inhibiting the initiation complex from forming - translation is therefore inhibited (see the figure below). In the dephosphorylated state eIF-2 can bind GTP and allow the assembly of the translation initiation complex as described above. The ability of the cell therefore to tune the assembly of the translation invitation complex via a reversible chemical modification (phosphorylation) to a regulatory protein is another example of how Nature has taken advantage of even this seemingly simple step to tuned gene expression.
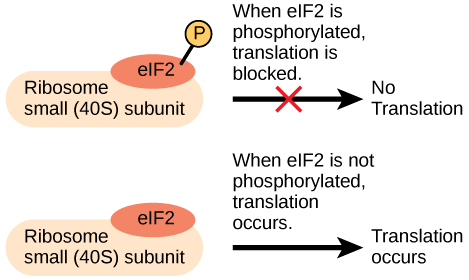
An increase in phosphorylation levels of eIF-2 has been observed in patients with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s. What impact do you think this might have on protein synthesis?
Chemical Modifications, Protein Activity, and Longevity
Not to be outdone by nucleic acids, proteins can also be chemically modified with the addition of groups including methyl, phosphate, acetyl, and ubiquitin groups. The addition or removal of these groups from proteins can regulate their activity or the length of time they exist in the cell. Sometimes these modifications can regulate where a protein is found in the cell—for example, in the nucleus, the cytoplasm, or attached to the plasma membrane.
Chemical modifications can occur in response to external stimuli such as stress, the lack of nutrients, heat, or ultraviolet light exposure. In addition to regulating the function of the proteins themselves, if these changes occur on specific proteins they can alter epigenetic accessibility (in the case of histone modification), transcription (transcription factors), mRNA stability (RNA binding proteins), or translation (eIF-2) thus feeding back and regulating various parts of the process of gene expression. In the case of modification to regulatory proteins, this can be an efficient way for the cell to rapidly change the levels of specific proteins in response to the environment by regulating various steps in the process.
The addition of an ubiquitin group has another function - it marks that protein for degradation. Ubiquitin is a small molecule that acts like a flag indicating that the tagged proteins should be targeted to an organelle called the proteasome. This organelle is a large multi-protein complex that functions to cleave proteins into smaller pieces that can then be recycled. Ubiquitination (the addition of a ubiquitin tag), therefore helps to control gene expression by altering the functional lifetime of the protein product.
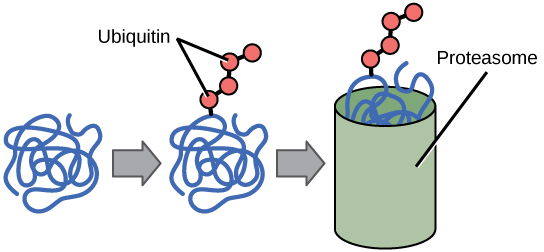
Proteins with ubiquitin tags are marked for degradation within the proteasome.
In conclusion, we see that gene regulation is complex and that it can be modulated at each step in the process of expressing a functional gene product. Moreover, the regulatory elements that happen at each step can act to influence other regulatory steps both earlier and later in the process of gene expression (i.e. the process of chemically altering a transcription factor can influence the regulation of its own transcription many steps earlier in the process). These complex sets of interactions form what are known as gene regulatory networks. Understanding the structure and dynamics of these networks is critical for understanding how different cells function, the basis for numerous diseases, developmental processes, and how cells make decisions about how to react to the many factors that are in constant flux both inside and outside.