
Spring_2024_Bis2A_Igo_Reading_06
- Page ID
- 132978
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Learning Objectives Associated with Spring_2024_Bis2A_Igo_Reading_06GC.38 Describe the concept of equilibrium in the context of reaction coordinate diagrams. GC.39 Interpret reaction coordinate diagrams and associate changes in Gibbs enthalpy and activation energy with relative rates of reactions, equilibrium conditions, and whether a reaction is endergonic or exergonic. GC.40 Understand how to use the equation ΔG = ΔH - TΔS and explain what each term represents. GC.41 Interpret a biochemical transformation and predict whether or not the reaction is spontaneous by using a Gibbs enthalpy (energy) reaction coordinate diagram. GC.46 Develop and apply an "energy story" about a biological or biochemical reaction using the first and second laws of thermodynamics. Describe events in terms of energy, awareness of energy conservation, energy transfer, entropy, and then relate these to what is happening at the molecular level. GC.47 Explain the first law of thermodynamics (conservation of energy). GC.48 Explain the second law of thermodynamics (entropy is increasing) and how it relates to biological reactions. GC.34 Interpret reaction coordinate diagrams showing either or both catalyzed and uncatalyzed reaction coordinates and identify respective activation energy barriers and relate these to the forward and reverse rates of reaction. GC.36 Describe mechanisms used by enzymes to lower the activation energy and increase rates of reaction. MS.18 Hypothesize how binding of small molecules to one or more binding pockets can lead to changes in protein function (i.e. competitive inhibition and/or allostery). MS.17 Draw a rough sketch of an enzyme including its active site and other sites in the enzyme that might impact its function, such as an inhibitor binding site. GC.42 Predict whether two reactions can be theoretically productively coupled by interpreting tables of standard Gibbs enthalpy (energy). GC.50 Describe why an enzyme is critical for coupling an exergonic reaction to an endergonic reaction by applying the concepts from the chemistry and thermodynamics lectures. MS. 14 Form hypotheses about how different types of changes at a molecular interface might influence binding (i.e. proteins-proteins, protein, DNA, protein-lipid, small molecule-lipid, small molecule-protein, etc.).This would include recognizing or inferring different types of chemical bonds from an analysis or knowledge of functional group interactions
|
Thermodynamics
Thermodynamics is concerned with describing the changes in systems before and after a change. This usually involves a discussion about energy transfers and its dispersion within the system and its surroundings. In nearly all practical cases, these analyses require that the system and its surroundings be completely described. For instance, when discussing the heating of a pot of water on the stove, the system may include the stove, the pot, and the water and the environment or surroundings may include everything else. Biological organisms are what are called open systems; energy is transferred between them and their surroundings.
The First Law of Thermodynamics
The first law of thermodynamics deals with the total amount of energy in the universe. It states that this total amount of energy is constant. In other words, there always has been, and always will be, exactly the same amount of energy in the universe.
According to the first law of thermodynamics, energy may be transferred from place to place, but it cannot be created or destroyed. Energy transfers take place around us all the time. Light bulbs transfer energy from electrical power stations into heat and photons of light. Gas stoves transfer energy stored in the bonds of chemical compounds into heat and light. (Heat, by the way, is the amount of energy transferred from one system to another because of a temperature difference.)
Plants perform one of the most biologically useful energy transfers on earth: they transfer energy in the photons of sunlight into the chemical bonds of organic molecules. In every one of these cases, energy is neither made nor destroyed, and we must try to account for all the energy when we examine some of these reactions.
The First Law and the Energy Story
The first law of thermodynamics is deceptively simple. Students often understand that energy cannot be created or destroyed. Yet, when describing an energy story of a process they often make the mistake of saying things such as "energy is produced from the transfer of electrons from atom A to atom B." While most of us will understand the point the student is trying to make, the wrong words are being used. Energy is not made or produced; it is simply transferred. To be consistent with the first law, when telling an energy story, make sure you try to track explicitly all the places that ALL the energy in the system at the start of a process goes by the end of a process.
The Second Law of Thermodynamics
An important concept in physical systems is entropy. Entropy relates to how energy can be distributed or dispersed within the particles of a system. The Second Law of Thermodynamics states that entropy is always increasing in a system and its surroundings (that is, everything inside and outside the system combined).
This idea helps explain the directionality of natural phenomena. The notion is that the directionality comes from the tendency for energy in a system to move towards a state of maximal dispersion. The Second Law, therefore, implies that in any transformation, we should look for an overall increase in entropy (or dispersion of energy), somewhere. As dispersion of energy in a system or its surroundings increases, the ability of the energy to be directed towards work decreases.
Keep in mind: you will find many examples in which the entropy of a system decreases locally. However, according to the Second Law, the entropy of the entire universe can never decrease. This must mean that there is an equal or greater increase in entropy somewhere else in the surroundings (most likely in a closely connected system) that compensates for the local decrease.
We associate the four scenarios below with increasing entropy of the system. Try to think of specific examples for when:
a. the system gains energy;
b. a change of state occurs from solid to liquid to gas;
c. a mixing of substances occurs;
d. the number of particles increases during a reaction.
Possible NB Discussion  Point
Point
Justify or refute the following statement: "Biological systems are an exception to the Second Law of Thermodynamics, since cells are known to arrange themselves into very highly organized structures (think: tissues, organs, etc.) rather than into a more disordered state." Make sure to check out what your peers are saying -- do you agree or disagree with their position and/or their rationale?

Figure 1. An increase in disorder can happen in different ways. An ice cube melting on a hot sidewalk is one example. Here, ice is displayed as a snowflake, with organized, structured water molecules forming the snowflake. Over time, the snowflake will melt into a pool of disorganized, freely moving water molecules. It is common to describe entropy as a measure of order as a way to simplify the more concrete description relating entropy to the number of states in which energy can be dispersed in a system. While the idea of measuring order to define entropy has some flaws, it is sometimes a useful, if imperfect, proxy. (Source)
If we consider the first and second laws together, we come to a useful conclusion. Whenever energy is transferred or redistributed within a system, entropy must increase. This increase in entropy is related to how "useful" the energy is to do work. Recall again that this energy becomes less and less available as entropy increases.
Keep in mind: you will find many examples in which the entropy of a system decreases locally. However, according to the Second Law, the entropy of the entire universe can never decrease. This must mean that there is an equal or greater increase in entropy somewhere else in the surroundings (most likely in a closely connected system) that compensates for the local decrease.
We conclude that while all the energy must be conserved, if the required change increases entropy, it means that some energy will become distributed in a way that makes it less useful for work. Most times, particularly in biology, some increase in entropy can be chalked up to a transfer of energy to heat in the environment.
The energy story
Overview of the energy story
Whether we know it, we tell stories that involve matter and energy every day. We just rarely use terminology associated with scientific discussions of matter and energy.
Example 1
The setup: a simple statement with implicit details
You tell your roommate a story about how you got to campus by saying, "I biked to campus today." In this simple statement are several assumptions that are instructive to unpack, even if they may not seem very critical to include explicitly in a casual conversation between friends over transportation choices.
An outsider's reinterpretation of the process
To illustrate this, imagine an external observer, such as an alien being watching the comings and goings of humans on Earth. Without the benefit of knowing much of the implied meanings and reasonable assumptions that are buried in our language, the alien's description of the morning cycling trip would differ from your own. What you described efficiently as "biking to campus" might be more specifically described by the alien as a change in location of a human body and its bicycle from one location (the apartment, termed position A) to a different location (the university, termed position B). The alien might be even more abstract and describe the bike trip as the movement of matter (the human body and its bike) between an initial state (at location A) to a final state (at location B). From the alien's standpoint, what you'd call "biking" might be more specifically described as the use of a two-wheeled tool that couples the transfer of energy from the electric fields in chemical compounds to the acceleration of the two-wheeled, tool-person combo that heats its environment. Finally, buried within the simple statement describing how we got to work is also the tacit understanding that the mass of the body and bike were conserved in the process (with some important caveats we’ll look at in future lectures) and that some energy was transferred around the system and environment to enable the movement of the body from position A to position B.
Details are important. What if you owned a fully electric bike, and the person you were talking with didn’t know that? What important details might this change about the “every day” story you told that the more detailed description would have cleared up? How would the alien’s story have changed? In what scenarios might these changes be relevant?
As this simple story illustrates, irrespective of many factors, the act of creating a full description of a process includes some accounting of what happened to the matter, what happened to the energy, and almost always some description of a mechanism that describes how changes in matter and energy of a system were brought about.
To practice this skill in BIS2A, we will make use of something we like to call the "Energy Story." You may be asked to tell an "energy story" in class, to practice telling energy stories on your lecture study guides, and to use the concept on your exams. In this section, we focus primarily on introducing the concept of an energy story and explaining how to tell one. It is worth noting that the term "energy story" is used almost exclusively in BIS2A (and has a specific meaning in this class). This precise term will not appear in other courses at UC Davis (at least in the short term), or if it appears, is not likely to be used in the same manner. You can think of “The Energy Story” as a systematic approach to creating a statement or story describing a biological process or event. Your BIS2A instructors have given this approach a short name (energy story) so that we can all associate it with the common exercise. That way, when the instructor asks the class to tell or construct an energy story, everyone knows what is meant.
Definition 1: energy story
An energy story is a narrative describing a process or event. The critical elements of this narrative are as follows:
- Identify at least two states (e.g., start and end) in the process.
- Identify and list the matter in the system and its state at the start and end of the process.
- Describe the transformation of the matter that occurs during the process.
- Account for the “location” of energy in the system at the start and end of the process.
- Describe the transfer of energy that happens during the process.
- Identify and describe mechanism(s) responsible for mediating the transformation of matter and transfer of energy.
A complete energy story will include a description of the initial reactants and their energetic states as well as a description of the final products and their energetic states after the process or reaction is completed.
Possible NB Discussion Point
We argue that the energy story can be used to communicate all the useful details that are required to describe nearly any process. Can you think of a process that cannot be adequately described by an energy story? If so, describe such a process.
Example 2: energy story example
Let us suppose that we are talking about the process of driving a car from "Point A" to "Point B" (see Figure 1).
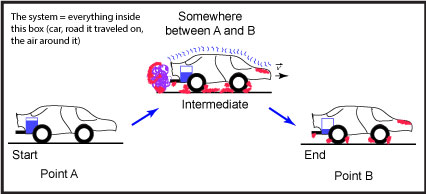
Figure 1: This is a schematic of a car moving from a starting position, "Point A," to an end point, "Point B." The blue rectangle depicted in the back of the car represents the level of the gasoline; the purple, squiggly line near the exhaust pipe represents the exhaust; squiggly blue lines on top of the car represent sound vibrations; and the red shading represents areas that are hotter than at the start. Source: created by Marc T. Facciotti (own work)
Let's step through the Energy Story rubric:
1. Identify at least two states (e.g., start and end) in the process.
In this example, we can easily identify two states. The first state is the nonmoving car at "Point A," the start of the trip. The second state, after the process is done, is the nonmoving car at "Point B."
2. Identify and list the matter in the system and its state at the start and end of the process.
In this case, we first note that the "system" includes everything in the figure—the car, the road, the air around the car, etc.
It is important to understand the we are going to apply the physical law of conservation of matter. That is, in any of the processes that we will discuss, matter is neither created or destroyed. It might change form, but one should be able to account for everything at the end of a process that was there at the beginning.
At the beginning of the process, the matter in the system consists of the following:
1. The car and all the stuff in it
2. The fuel in the car (a special thing in the car)
3. The air (including oxygen) around the car.
4. The road
5. The driver
At the end of the process, the matter in the system is distributed as follows:
1. The car and all the stuff in it is in a new place (let's assume, aside from the fuel and position, that nothing else changed).
2. There is less fuel in the car, and it too is in a new place.
3. The air has changed; it now has less molecular oxygen, more carbon dioxide, and more water vapor.
4. The road did not change (let's assume it didn't change—other than a few pebbles that moved around).
5. The driver did not change (let's assume she didn't change—though we'll see by the end of the term that she did, at least a little). However, the driver is now in a different place.
3. Describe the transformation of the matter that occurs during the process.
What happened to the matter in this process? Thanks to a lot of simplifying assumptions, we see that two big things happened. First, the car and its driver changed positions—they went from "Point A" to "Point B." Second, we note that some of the molecules in the fuel, which used to be in the car as a liquid, have changed forms and are now mostly in the form of carbon dioxide and water vapor (purple blob coming out of the tailpipe). Some of the oxygen molecules that used to be in the air are now also in a new place as part of the carbon dioxide and water that left the car.
4. Account for the “location” of energy in the system at the start and end of the process.
It is again important to understand that we are going to invoke the physical law of conservation of energy. That is, we stipulate that the energy in the system cannot be created or destroyed, and therefore, the energy that is in the system at the start of the process must still be there at the end of the process. It may have been redistributed, but you should be able to account for all the energy.
At the beginning of the process, the energy in the system is distributed as follows:
1. The energy is tied up in the associations between atoms that make up the matter of the car.
2. The energy is tied up in the associations between atoms that make up the fuel.
3. The energy is tied up in the associations between atoms that make up the air.
4. The energy is tied up in the associations between atoms that make up the road.
5. The energy is tied up in the associations between atoms that make up the driver.
6. For all the things above, we can also say that there is energy in the molecular motions of the atoms that make up the stuff.
At the end of the process, the energy in the system is distributed as follows:
1. The energy is tied up in the associations between atoms that make up the matter of the car.
2. The energy is tied up in the associations between atoms that make up the fuel.
3. The energy is tied up in the associations between atoms that make up the air.
4. The energy is tied up in the associations between atoms that make up the road.
5. The energy is tied up in the associations between atoms that make up the driver.
6. For all the things above, we can also say that there is energy in the molecular motions of the atoms that make up the stuff.
This is interesting in some sense, because the lists are about the same. We know that the amount of energy stored in the car has decreased, because there is less fuel. Something must have happened.
5. Describe the transfer of energy that happens during the process.
In this particular example, it is the transfer of energy among the components of the system that is most interesting. As we mentioned, there is less energy stored in the gas tank of the car at the end of the trip, because there is now less fuel. We also know intuitively (from real-life experience) that the transfer of energy from the fuel to something else was instrumental in moving the car from "Point A" to "Point B." So, where did this energy go? Remember, it didn't just disappear. It must have moved somewhere else in the system.
Well, we know that there is more carbon dioxide and water vapor in the system after the process. There is energy in the associations between those atoms (atoms that used to be in the fuel and air). So some of the energy that was in the fuel is now in the exhaust. Let's also draw from real-life experience again, and state that we know that parts of our car have gotten hot by the end of the trip (e.g., the engine, transmission, wheels/tires, exhaust, etc.). For the moment, we'll just use our intuition, and say that we understand that making something hot involves some transfer of energy. So we can reasonably postulate that some of the energy in the fuel went (directly or indirectly) into heating the car, parts of the road, and the exhaust—and thus the environment around the car. An amount of energy also went into accelerating the car from zero velocity to whatever speed it traveled at, but most of that energy eventually became heat when the car came to a stop.
This is a bit of a hand-wavy explanation, and we'll learn how to do a better job throughout the quarter. The main point is that we should be able to add all the energy of the system at the beginning of the process (in all the places it is found) and at the end of the process (in all the places it is found), and those two values should be the same.
6. Identify and describe mechanism(s) responsible for mediating the transformation of matter and transfer of energy.
Finally, it is useful to try understanding how those transformations of matter and transfers of energy might have been facilitated. For the sake of brevity, we might just say that there was a complicated mechanical device (the engine) that helped facilitate the conversion of matter and transfer of energy about the system and coupled this to the change in position of the car. Someone interested in engines would, of course, give a more detailed explanation.
In this example, we made a bunch of simplifying assumptions to highlight the process and to focus on the transformation of the fuel. But that's fine. The more you understand about the processes, the finer details you can add. Note that you can use the Energy Story rubric for describing your understanding (or looking for holes in your understanding) of nearly any process (certainly in biology). In BIS2A, we'll use the Energy Story to get an understanding of processes as varied as biochemical reactions, DNA replication, the function of molecular motors, etc.
Important:
First: We will be working on many examples of the energy story throughout the course—do not feel that you need to have mastery over this topic today.
Second: While it is tempting to think all this is superfluous or not germane to your study of biology in BIS2A, let this serve as a reminder that your instructors (those creating the course midterm and final assessments) view it as core material. We will revisit this topic often throughout the course but need you to get familiar with some of the basic concepts now.
This is important material and an important skill to develop—do not put off studying it because it doesn't "look" like "biology" to you today. The academic term moves VERY quickly, and it will be difficult to catch up later if you don't give this some thought now.
Energy
Energy is a central concept in all sciences. Energy is a property of a system. Understanding the transfer of energy around physical systems is a key component of understanding how and why things change. In the following sections, we will explore some basic concepts associated with common transformations in biology and chemistry: the solubility of various biomolecules, the making and breaking of chemical bonds, transferring electrons, transferring energy to and from light, and transferring energy as heat. In class, many of the discussions will happen in the context of the Energy Story rubric, so when we consider a reaction of transformation, we will be interested in precisely defining the system in question and trying to account for all the various transfers of energy that occur within the system, making sure that we abide by the Law of Conservation of Energy.
There are plenty of examples where we use the concept of energy in our everyday lives to describe processes. A bicyclist can bike to get to campus for class. The act of moving herself and her bicycle from point A to point B can
How we will approach conceptualizing energy
In BIS2A we will think about energy with a "stuff" metaphor. Note, however, that energy is NOT a substance, it is rather a property of a system. But we will think of it, in some sense, as property that can
Energy sources
Ultimately, the source of energy for many processes occurring on the Earth's surface comes from solar radiation. But as we will see, biology has been very clever at tapping a variety of forms of energy to construct and maintain living beings. As we move through this course, we will explore a variety of energy sources and the ways in which biology has devised to transfer energy from these fuels.
Energy in chemical reactions
Chemical reactions involve a redistribution of energy within the reacting chemicals and with their environment. So, like it or not, we need to develop some models that can help us describe where energy is in a system (perhaps how it is "stored"/distributed) and how it can be moved around in a reaction. The models we develop will not be overly detailed in the sense that they would satisfy a hard-core chemist or physicist with their level of technical detail, but we expect that they should still be technically correct and not form incorrect mental models that will make it difficult to understand the "refinements" later.
In this respect, one of the key concepts to understand is that we will think about energy being transferred between parts of a system rather than referring thinking too much about it as being transformed. The distinction between "transfer" and "transform" is important because the latter gives the impression that energy is a property that exists in different forms, that it gets reshaped somehow. The common use of the term "transform" in relation to energy is understandable as different phenomena associated with the concept of energy physically "look" different to us. However, one potential problem with using the "transform" language is that it is sometimes difficult to reconcile with the idea that energy is being conserved (according to the first law of thermodynamics) if it is constantly changing form. How can the entity of energy be conserved if after a transformation it is no longer the same thing (e.g. transformed)? The second law of thermodynamics tells us that no transformation conserves all energy in a system. If energy is getting "transformed," how can it be conserved and still be consistent with the second law of thermodynamics?
So, instead, we will approach this issue by transferring and storing energy between different parts of a system and thus think about energy as a property that can get redistributed. That'll hopefully make the accounting of energy easier. Not that the energy "transfer" idea is consistent and compatible with terms like "potential energy" and "kinetic energy", as these are useful for describing how the energy is distributed between the motion of matter and the various fields (e.g. electric, gravitational, etc.) in a system.
CAUTION
If we are going to think about transferring energy from one part of a system to another, we also need to be careful about NOT treating energy like a substance that moves like a fluid or "thing." Rather, we need to appreciate energy simply as a property of a system that can be measured and reorganized but that is neither a "thing" nor something that is at one time in one form then later in another.
Since we will often be dealing with transformations of biomolecules, we can start by thinking about where energy can be found/stored in these systems. We'll start with a couple of ideas and add more to them later.
Let us propose that one place that energy can be stored is in the motion of matter. For brevity, we'll give the energy stored in motion a name: kinetic energy. Molecules in biology are in constant motion and therefore have a certain amount of kinetic energy (energy stored in motion) associated with them.
Let us also propose that there is a certain amount of energy stored in the biomolecules themselves and that the amount of energy stored in those molecules is associated with the types and numbers of atoms in the molecules and their organization (the number and types of bonds between them). The discussion of exactly where the energy is stored in the molecules is beyond the scope of this class, but we can approximate it by suggesting that a good proxy is in the bonds. Different types of bonds may be associated with storing different amounts of energy. In some contexts, this type of energy storage could be labeled potential energy or chemical energy. With this view, one of the things that happens during the making and breaking of bonds in a chemical reaction is that the energy is transferred about the system into different types of bonds. In the context of an Energy Story, one could theoretically count the amount of energy stored in the bonds and motion of the reactants and the energy stored in the bonds and energy of the products.
In some cases, you might find that when you add up the energy stored in the products and the energy stored in the reactants that these sums are not equal. If the energy in the reactants is greater than that in the products, where did this energy go? It had to get transferred to something else. Some will certainly have moved into other parts of the system, stored in the motion of other molecules (warming the environment) or perhaps in the energy associated with photons of light. One good, real-life example is the chemical reaction between wood and oxygen (reactants) and it's conversion to carbon dioxide and water (products). At the beginning, the energy in the system is largely in the molecular bonds of oxygen and the wood (reactants). There is still energy left in the carbon dioxide and water (products) but less than at the beginning. We all appreciate that some of that energy was transferred to the energy in light and heat. This reaction where energy is transferred to the environment is termed exothermic. By contrast, in some reactions, energy will transfer in from the environment. These reactions are endothermic.
The transfer of energy in or out of the reaction from the environment is NOT the only thing that determines whether a reaction will be spontaneous or not. We'll discuss that soon. For the moment, it is important to get comfortable with the idea that energy can be transferred among different components of a system during a reaction and that you should be able to envision tracking it.
Free Energy

If we want to describe transformations, it is useful to have a measure of (a) how much energy is in a system, (b) the dispersal of that energy within the system and (c) how these factors change between the start and end of a process. The concept of free energy, often referred to as Gibbs energy or Gibbs enthalpy (abbreviated with the letter G), in some sense, does just that. We can define Gibbs energy in several interconvertible ways, but a useful one in the context of biology is the enthalpy (internal energy) of a system minus the entropy of the system scaled by the temperature. The difference in free energy when a process takes place is often reported in terms of the change (Δ) of enthalpy (internal energy) denoted H, minus the temperature scaled change (Δ) in entropy, denoted S. See the equation below.
\[ΔG=ΔH−TΔS \label{gibbs}\]
We often interpret the Gibbs energy as the amount of energy available to do useful work. With a bit of hand waving, we can interpret this statement by invoking the idea presented in the section on entropy, which states the dispersion of energy (required by the Second Law) associated with a positive change in entropy somehow renders some energy that is transferred less useful to work. One can say that it reflects this in part in the \(T∆S\) term of the Equation \ref{gibbs}.
To provide a basis for fair comparisons of changes in Gibbs energy amongst different biological transformations or reactions, the free energy change of a reaction is measured under a set of common standard experimental conditions. The resulting standard free energy change of a chemical reaction is expressed as an amount of energy per mole of the reaction product (either in kilojoules or kilocalories, kJ/mol or kcal/mol; 1 kJ = 0.239 kcal), when measured at a standard pH, temperature, and pressure conditions. Standard pH, temperature, and pressure conditions are generally standardized at pH 7.0, 25 degrees Celsius, and 100 kilopascals (1 atm pressure), respectively. It is important to note that cellular conditions differ from these standard conditions, and so actual \(∆G\) inside a cell will differ considerably from those calculated under standard conditions.
Chemical Equilibrium—Part 2: Gibbs Energy
In a previous section, we began a description of chemical equilibrium in the context of forward and reverse rates. We presented three key ideas:
- At equilibrium, the concentrations of reactants and products in a reversible reaction are not changing in time.
- A reversible reaction at equilibrium is not static—reactants and products continue to interconvert at equilibrium, but the rates of the forward and reverse reactions are the same.
- We were NOT going to fall into a common student trap of assuming that chemical equilibrium means that the concentrations of reactants and products are equal at equilibrium.
Here we extend our discussion and put the concept of equilibrium into the context of Gibbs energy, also reinforcing the Energy Story exercise of considering the "Before/Start" and "After/End" states of a reaction (including the inherent passage of time).
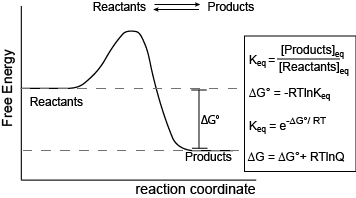
Figure 1. Reaction coordinate diagram for a generic exergonic reversible reaction. Equations relating Gibbs energy and the equilibrium constant: R = 8.314 J mol-1 K-1 or 0.008314 kJ mol-1 K-1; T is temperature in Kelvin. Attribution: Marc T. Facciotti (original work)
The figure above shows a commonly cited relationship between ∆G° and Keq:
\[ ∆G^o = -RT\ln K_{eq}.\]
Here, G° indicates the Gibbs energy under standard conditions (e.g., 1 atmosphere of pressure, 298 K). This equation describes the change in Gibbs energy for reactants converting to products in a reaction that is at equilibrium. The value of ∆G° can therefore be thought of as being intrinsic to the reactants and products themselves. ∆G° is like a potential energy difference between reactants and products. With this concept as a basis, one can also consider a reaction where the "starting" state is somewhere out of equilibrium. In this case, there may be an additional “potential” associated with the out-of-equilibrium starting state. This “added” component contributes to the ∆G of a reaction and can be effectively added to the expression for Gibbs energy as follows:
\[∆G = ∆G° + RT\ln Q, \]
where \(Q\) is called the reaction quotient. From the standpoint of General Biology, we will use a simple (a bit incomplete but functional) definition for
\[Q = \dfrac{[Products]_{st}}{[Reactants]_{st}} \]
at a defined non-equilibrium condition, st. One can extend this idea and calculate the Gibbs energy difference between two non-equilibrium states, provided they are properly defined and thus compute Gibbs energy changes between specifically defined out-of-equilibrium states. This last point is often relevant in reactions found in biological systems as these reactions are often found in multi-step pathways that effectively keep individual reactions in an out-of-equilibrium state.
This takes us to a point of confusion for some. In many biology books, the discussion of equilibrium includes not only the discussion of forward and reverse reaction rates, but also a statement that ∆G = 0 at equilibrium. This can be confusing because these very discussions often follow discussions of non-zero ∆G° values in the context of equilibrium (∆G° = -RTlnKeq). The nuance to point out is that ∆G° is referring to the Gibbs energy potential inherent in the chemical transformation between reactants and products alone. This is different from considering the progress of the reaction from an out-of-equilibrium state that is described by
\[∆G = ∆G^o + RT \ln Q.\]
This expression can be expanded as follows:
\[∆G = -RT\ln K_{eq} + RT\ln Q\]
to bring the nuance into clearer focus. In this case note that as Q approaches Keq that the reaction ∆G becomes closer to zero, ultimately reaching zero when Q = Keq. This means that the Gibbs energy of the reaction (∆G) reaches zero at equilibrium not that the potential difference between substrates and products (∆G°) reaches zero.
Endergonic and exergonic reactions
Any system of molecules that undergoes a physical transformation/reorganization (aka. reaction) will have an associated change in internal energy and entropy. If we examine a single isolated reaction, in which unique reactants convert into unique products the Gibbs energy of the system will be dependent several factors, key among which are (a) the internal energy and entropy differences associated with the molecular rearrangements and (b) the degree to which the reaction is out-of-equilibrium.
If, for the sake of simplicity we begin by considering only the contribution of the molecular transformations in the system on ∆G, we conclude that reactions with ∆G < 0, the products of the reaction have less Gibbs energy than the reactants. Since ∆G is the difference between the enthalpy and temperature-scaled entropy changes in a reaction, a net negative ∆G can arise in through changes largely of enthalpy, entropy or most often both. The left panel of Figure 1 below shows a common graphical representation of an exergonic reaction. This graph is called a reaction coordinate diagram. It plots Gibbs energy on the y-axis, and the x-axis in arbitrary units shows the progress of a reaction. With an exergonic reaction, the figure on the left shows two key things: (1) the difference between the free energy of the reactants and products is negative and (2) the progress of the reaction requires some input of free energy (shown as an energy "hill" or barrier). This graph does not tell us how the energy in the system redistributed, only that the difference between enthalpy and temperature-scaled entropy is negative. Exergonic reactions are said to occur spontaneously. Understanding which chemical reactions are spontaneous is useful for biologists who are trying to understand whether a reaction is likely to "go" or not.
It is important to note that the term "spontaneous"—in thermodynamics—implies nothing about how fast the reaction proceeds. The change in free energy only describes the difference between beginning and end states, NOT how fast that transition takes place. This is contrary to the everyday use of the term which usually carries the implicit understanding that something happens quickly. As an example, the oxidation/rusting of iron is a spontaneous reaction. However, an iron nail exposed to air does not rust instantly—it may take years.
A chemical reaction with a positive ∆G means that the products of the reaction have a higher free energy than the reactants (see the right panel of Figure 1). These chemical reactions are called endergonic reactions, and they are NOT spontaneous. An endergonic reaction will not take place on its own without transferring energy into the reaction or increase of entropy somewhere else.
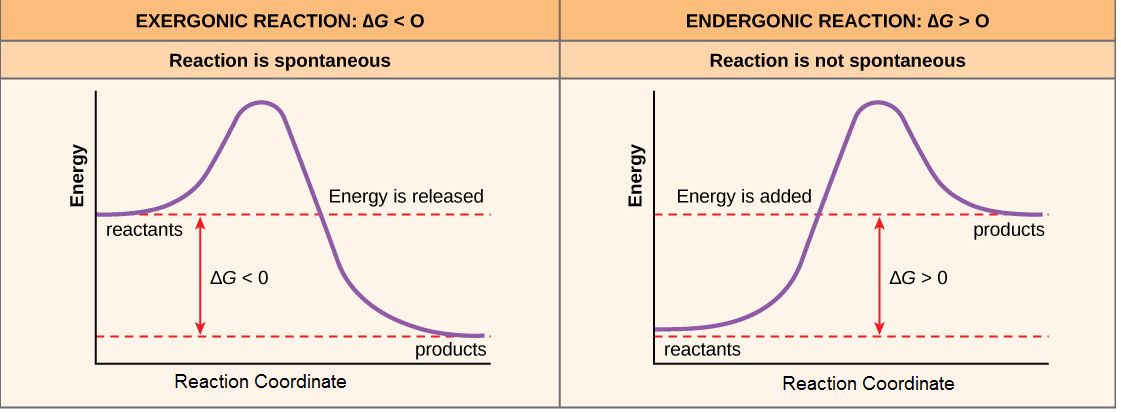
Figure 1. Reaction coordinate diagrams of exergonic and endergonic reactions. Exergonic and endergonic reactions are characterized by changes in Gibbs energy. In the equilibrium state of an exergonic reaction, the Gibbs energy of the products is lower than that of the reactants. Meanwhile, the equilibrium state of an endergonic reaction in, the Gibbs energy of the products is higher than that of the reactants. Attribution: Marc T. Facciotti (own work)
The building of complex molecules, such as sugars, from simpler ones is an anabolic process and is endergonic. On the other hand, the catabolic process, such as the breaking down of sugar into simpler molecules, is exergonic. Like the example of rust above, while the breakdown of biomolecules is generally spontaneous, these reactions don’t occur need to occur instantaneously (quickly). Remember, the terms endergonic and exergonic only refer to the difference in Gibbs energy between the products and reactants; they don't tell you about the rate of the reaction (how fast it happens). Rate will be discussed in later sections.
An important concept in the study of metabolism and energy is that of chemical equilibrium. Most chemical reactions are reversible. They can proceed in both directions, often transferring energy into their environment in one direction and transferring energy in from the environment in the other direction. The same is true for the chemical reactions involved in cell metabolism, such as the breaking down and building up of proteins into and from individual amino acids, respectively. Reactants within a closed system will undergo chemical reactions in both directions until a state of equilibrium is reached. Equilibrium in a chemical reaction is the state in which both reactants and products are present in concentrations that have no further tendency to change with time. Usually, this state results when the forward reaction proceeds at the same rate as the reverse reaction. NOTE THIS LAST STATEMENT! Equilibrium means that the relative concentrations of reactants and products are not changing in time, BUT it does NOT mean that there is no interconversion between substrates and products—it just means that when the reactant(s) are converted to product(s) that product(s) are converted to reactant(s) at an equal rate (see Figure 2). The state of equilibrium is also one of the lowest possible free energy states for the reaction and is a state of maximal entropy.
If a reaction is kept or started far out of equilibrium this state of the system also contributes to the overall Gibbs energy of a reaction. Either a rebalancing of substrate or product concentrations (by adding or removing substrate or product) or a positive change in free energy, typically by the transfer of energy from outside the reaction, can to move a reaction to an out-of-equilibrium state. Note that in a living cell, most chemical reactions do not reach a state of equilibrium—this would require that they reach their lowest free energy state, a state that is almost by definition incompatible with life. Energy is therefore required to keep biological reactions out of their equilibrium state. In this way, living organisms are in a constant, energy-requiring, uphill battle against equilibrium and entropy. This also means that the Gibbs energy of most biological reactions as they occur in the cell must also include a contribution from this out-of-equilibrium state. The Gibbs energy of these reactions, therefore, is often different from that reported under standard conditions.
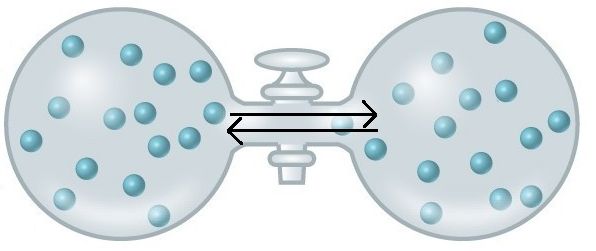
Figure 2. At equilibrium, do not think of a static, unchanging system. Instead, picture molecules moving in equal amounts from one area to another. Here, at equilibrium, molecules are still moving from left to right and right to left. The net movement, however, is equal. There will still be about 15 molecules in each side of this flask once it reaches equilibrium. Source: https://courses.candelalearning.com/...apter/entropy/
Catalysts
For a chemical reaction to happen, the reactants must first find one another in space. Chemicals in solution don't "plan" these collisions; they happen at random. In fact, most times, it's even more complicated. Not only do the reactants need to run into one another, but they also need to come into contact in a specific orientation. If reactants are very dilute, the rate of the reaction will be slow—collisions will happen infrequently. Increasing the concentrations will increase the rate of productive collisions. Another way to change the rate of reaction is to increase the rate of collisions by increasing the rate at which the reactants explore the reaction space—by increasing the velocity of the molecules or their kinetic energy. This can be accomplished by transferring heat into the system or raising the temperature. Those two strategies are often suitable for increasing the rates of chemical reactions that happen in a tube. However, in the cell, transferring heat may not be practical, as it may damage cellular components and lead to death. Cells sometimes use mechanisms to increase concentrations of reactants (we'll see some examples below), but this is rarely enough to drive reaction rates in a biologically relevant regime. This is where catalysts come in.
A catalyst is something that helps increase the rate of a chemical reaction undergoing no change itself. You can think of a catalyst as a chemical change agent.
The most important catalysts in biology are called enzymes. An enzyme is a protein catalyst. Other cellular catalysts include molecules called ribozymes. A ribozyme is a catalyst composed of a ribonucleic acid (RNA). Both will be discussed in more detail later in the course. Like all catalysts, enzymes work by lowering the level of energy that needs to be transferred into a chemical reaction to make it happen. A chemical reaction’s activation energy is the “threshold” level of energy needed to start the reaction.
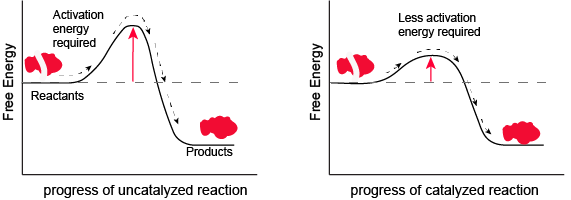
Figure 1. Enzymes and other catalysts decrease the activation energy required to start a given chemical reaction. Without an enzyme (left), the energy input needed for a reaction to begin is high. With the help of an enzyme (right), la reaction needs less energy to begin. Attribution: Marc T. Facciotti (original work)
In the figure above, What do you think the units are on the x-axis? Time would be one guess. However, if you compare the figures, it appears that the products are formed at the same time whether the activation energy barrier is high or low. Wasn't the point of this figure to illustrate that reactions with high activation energy barriers are slower than those with low activation energy barriers? What's going on?
Enzymes Section Overview
Enzymes are biological catalysts that speed up chemical reactions by lowering the activation energy. Enzymes are proteins comprising one or more polypeptide chains. Enzymes have an active site that provides a unique chemical environment made up of certain amino acid R groups (residues). This unique environment is well suited to convert particular chemical reactants for that enzyme, called substrates, into unstable intermediates, called transition states. Enzymes and substrates are thought to bind with an induced fit which means that enzymes and substrates undergo slight conformational adjustments upon substrate contact, leading to binding. Enzymes bind to substrates and catalyze reactions in four different ways: bringing substrates together in an optimal orientation, compromising the bond structures of substrates so that bonds can be more easily broken, providing optimal environmental conditions for a reaction to occur, or taking part directly in their chemical reaction by forming transient covalent bonds with the substrates.
Enzyme action must be regulated so that, in a given cell at a given time, the desired reactions are being catalyzed and the undesired reactions are not. Enzymes are regulated by cellular conditions, such as temperature and pH. They are also regulated through their location within a cell, sometimes being compartmentalized so that they can only catalyze reactions under certain circumstances. Inhibition and activation of enzymes via other molecules are other important ways that enzymes are regulated. Inhibitors can act competitively, noncompetitively, or allosterically; noncompetitive inhibitors are usually allosteric. Activators can also enhance the function of enzymes allosterically. The most common method by which cells regulate the enzymes in metabolic pathways is through feedback inhibition. During feedback inhibition, the products of a metabolic pathway serve as inhibitors (usually allosteric) of one or more of the enzymes (usually the first committed enzyme of the pathway) involved in the pathway that produces them.
Enzymes
A substance that helps a chemical reaction to occur is a catalyst, and the special molecules that catalyze biochemical reactions are called enzymes. Almost all enzymes are proteins, made up of chains of amino acids, and they perform the critical task of lowering the activation energies of chemical reactions inside the cell. Enzymes do this by binding to the reactant molecules and holding them in such a way as to make the chemical bond-breaking and bond-forming processes take place more readily. It is important to remember that enzymes don’t change the ∆G of a reaction. They don’t change whether a reaction is exergonic (spontaneous) or endergonic (not spontaneous). This is because they don’t change the free energy of the reactants or products. They only reduce the activation energy required to reach the transition state.
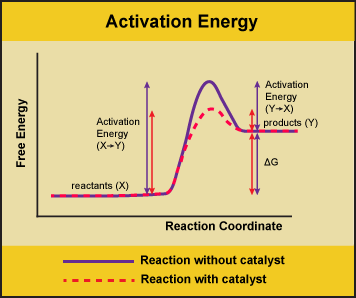
Figure 1. Enzymes lower the activation energy of the reaction but do not change the free energy of the reaction. Here, the solid line in the graph shows the energy required for reactants to turn into products without a catalyst. The dotted line shows the energy required using a catalyst. This figure should say Gibbs Free Energy on the Y-axis and instead of noting ∆H should have ∆G. Attribution: Marc T. Facciotti (own work)
Enzyme active site and substrate specificity
The chemical reactants to which an enzyme binds are the enzyme’s substrates. There may be one or more substrates, depending on the particular chemical reaction. In some reactions, a single-reactant substrate is broken down into multiple products. In others, two substrates may come together to create one larger molecule. Two reactants might also enter a reaction, both become changed, and leave the reaction as two products. The location within the enzyme where the substrate binds is called the enzyme’s active site. The active site is where the “action” happens, so to speak. Since enzymes are proteins, there is a unique combination of amino acid residues (also called side chains, or R groups) within the active site. Each amino acid side chain is characterized by different properties. Amino acids can be classified as large or small, weakly acidic or basic, hydrophilic or hydrophobic, positively or negatively charged, or neutral. The unique combination of amino acids (their positions, sequences, structures, and properties) creates a very specific chemical environment within the active site. This specific environment is suited to bind, albeit briefly, to a specific chemical substrate (or substrates). Due to this jigsaw-puzzle-like match between an enzyme and its substrates (which adapts to find the best fit between the transition state and the active site), enzymes are known for their specificity. The “best fit” between an enzyme and its substrates results from their respective shapes and the chemical complementarity of the functional groups on each binding partner.
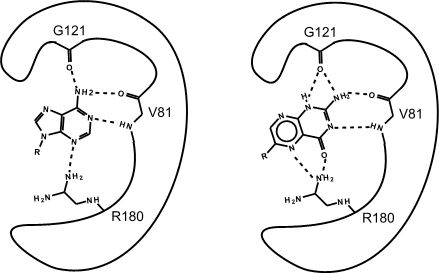
Figure 2. This is an enzyme with two different substrates bound in the active site. The enzymes are represented as blobs, except for the active site, which shows the three R-groups of each of the three amino acids in the active site. These R groups are interacting with the substrates through hydrogen bonding (represented as dashed lines).
At this point in the class, you should be familiar with all the types of bonds as well as the chemical characteristics of all the functional groups. For example, the R group of R180 in the enzyme depicted above is the amino acid Arginine (abbreviated as R) and has an R group that comprises several amino functional groups. Amino functional groups contain a nitrogen (N) and hydrogen (H) atoms. Nitrogen is more electronegative than hydrogen, so the covalent bond between N-H is a polar covalent bond. The hydrogen atoms in this bond will have a positive dipole moment, and the nitrogen atom will have a negative dipole moment. This allows amino groups to form hydrogen bonds with other polar compounds. Likewise, the backbone carbonyl oxygens of valine (V) 81 and glycine (G) 121 the backbone amino hydrogen of V81 are depicted engaged in hydrogen bonds with the small molecule substrate.
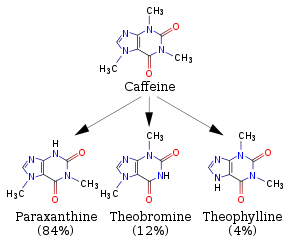
Possible NB Discussion Point: How your body breaks down caffeine
When you drink coffee or other caffeinated beverages like some sodas, you are consuming a molecule called caffeine! Caffeine over time gets metabolized (broken down) via a set of very related "CYP (Cytochrome P450)" enzymes to yield the three products shown in the figure below (Source: Wikipedia). To simplify slightly, you can interpret one arrow to represent a reaction catalyzed by one of the related CYP enzymes to yield paraxanthine, theobromine, or theophylline... all of which themselves get recognized by other enzymes that will further break them down and so on and so forth. Take a moment to examine the four structures below; the general structure should look vaguely familiar to you. Compare the reactant and the three products -- what are the noteworthy functional groups and properties of these molecules? What do you predict to be the key features of the active sites for the enzymes that break down these four molecules? If you were to design an enzyme that would break down caffeine AND theophylline only, how would you design your active site?
Exercise
Look to see which atoms in Figure 2 (above) are involved in the hydrogen bonds between the amino acid R groups and the substrate. You will need to be able to identify these on your own; hydrogen bonds may not be drawn in for you on the test.
If you changed the pH of the solution that this enzyme is located in, would the enzyme still be able to form hydrogen bonds with the substrate?
Which substrate (the left or right one) do you think is more stable in the active site? Why? How?
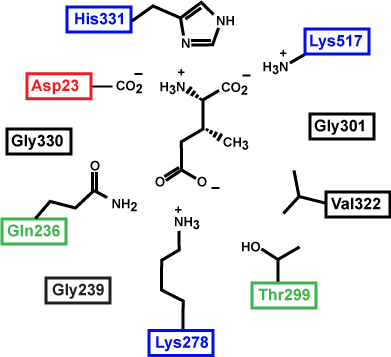
Figure 3. This is a depiction of an enzyme active site. Only the amino acids in the active site are drawn. The substrate is sitting directly in the center.
Source: created by Marc T. Facciotti (original work)
Exercise
First, identify the type of macromolecule in Figure 3. Second, draw in and label the appropriate interactions between the R groups and the substrate. Explain how these interactions might change if the pH of the solution changed.
Real-Life Connection
A new way of visualizing diseases in the body.
Structural instability of enzymes
The fact that active sites are so well suited to provide specific environmental conditions also means that they are subject to influences by the local environment. It is true that increasing the environmental temperature generally increases reaction rates, enzyme-catalyzed or otherwise. However, increasing or decreasing the temperature outside of an optimal range can affect chemical bonds within the active site in such a way that they are less well suited to bind substrates. High temperatures will eventually cause enzymes, like other biological molecules, to denature, a process that changes the natural properties of a substance. Likewise, the pH of the local environment can also affect enzyme function. Active site amino acid residues have their own acidic or basic properties that are optimal for catalysis. These residues are sensitive to changes in pH that can impair the way substrate molecules bind. Enzymes are suited to function best within a certain pH range, and, as with temperature, extreme pH values (acidic or basic) of the environment can cause enzymes to denature.
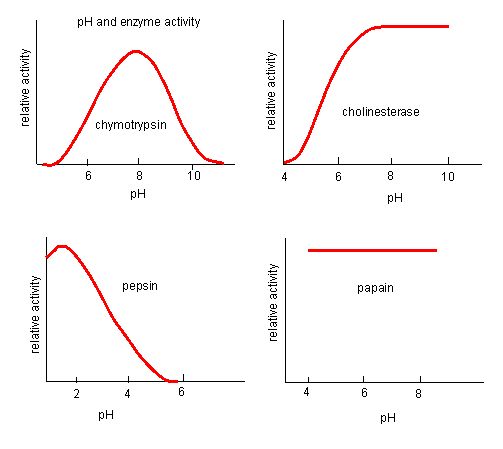
Figure 4. Enzymes have an optimal pH. The pH at which the enzyme is most active will be the pH where the active site R groups are protonated/deprotonated such that the substrate can enter the active site and the initial step in the reaction can begin. Some enzymes require a very low pH (acidic) to be completely active. In the human body, these enzymes are most likely located in the lower stomach, or located in lysosomes (a cellular organelle used to digest large compounds inside the cell).
Source: http://biowiki.ucdavis.edu/Biochemis..._pH_Inhibition
The process where enzymes denature usually starts with the unwinding of the tertiary structure through destabilization of the bonds holding the tertiary structure together. Hydrogen bonds, ionic bonds, and covalent bonds (disulfide bridges and peptide bonds) can all be disrupted by large changes in temperate and pH. Using the chart of enzyme activity and temperature below, make an energy story for the red enzyme. Explain what might be happening from 37 °C to 95 °C.
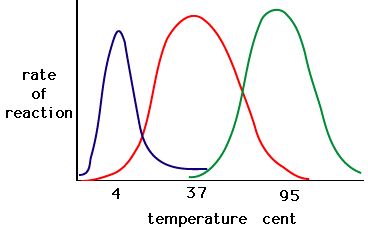
Figure 5. Enzymes have an optimal temperature. The temperature at which the enzyme is most active will usually be the temperature where the structure of the enzyme is stable or uncompromised. Some enzymes require a specific temperature to remain active and not denature. Source: http://academic.brooklyn.cuny.edu/bi...ge/enz_act.htm
Induced fit and enzyme function
For many years, scientists thought that enzyme-substrate binding took place in a simple “lock-and-key” fashion. This model asserted that the enzyme and substrate fit together perfectly in one instantaneous step. However, current research supports a more refined view called induced fit. The induced-fit model expands upon the lock-and-key model by describing a more dynamic interaction between enzyme and substrate. As the enzyme and substrate come together, their interaction causes a mild shift in the enzyme’s structure that confirms a more productive binding arrangement between the enzyme and the transition state of the substrate. This energetically favorable binding maximizes the enzyme’s ability to catalyze its reaction.
When an enzyme binds its substrate, an enzyme-substrate complex is formed. This complex lowers the activation energy of the reaction and promotes its rapid progression in one of many ways. On a basic level, enzymes promote chemical reactions that involve more than one substrate by bringing the substrates together in an optimal orientation. The appropriate region (atoms and bonds) of one molecule is juxtaposed to the appropriate region of the other molecule with which it must react. Another way in which enzymes promote the reaction of their substrates is by creating an energetically favorable environment within the active site for the reaction to occur. Certain chemical reactions might proceed best in a slightly acidic or nonpolar environment. The chemical properties that emerge from the particular arrangement of amino acid residues within an active site create the energetically favorable environment for an enzyme’s specific substrates to react.
The activation energy required for many reactions includes the energy involved in slightly contorting chemical bonds so that they can more easily react. Enzymatic action can aid this process. The enzyme-substrate complex can lower the activation energy by contorting substrate molecules in such a way as to facilitate bond breaking. Finally, enzymes can also lower activation energies by taking part in the chemical reaction itself. The amino acid residues can provide certain ions or chemical groups that actually form covalent bonds with substrate molecules as a necessary step of the reaction process. In these cases, it is important to remember that the enzyme will always return to its original state at the completion of the reaction. One of the hallmark properties of enzymes is that they remain ultimately unchanged by the reactions they catalyze. After an enzyme is done catalyzing a reaction, it releases its product(s).
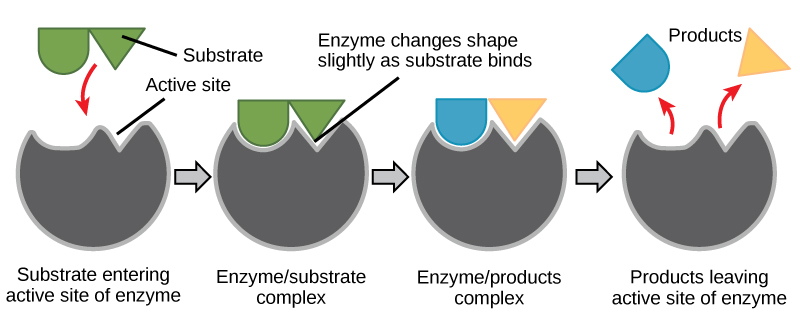
Figure 6. According to the induced-fit model, both enzyme and substrate undergo dynamic conformational changes upon binding. The enzyme contorts the substrate into its transition state, thereby increasing the rate of the reaction.
Creating an energy story for the reaction above
Using Figure 6, answer the questions posed in the energy story.
1. What are the reactants? What are the products?
2. What work was accomplished by the enzyme?
3. What state is the energy in initially? What state is the energy transformed into in the final state? This one might be tricky still, but try to identify where the energy is in the initial state and the final state.
Enzyme regulation
Why regulate enzymes?
Cellular needs and conditions vary from cell to cell and change within individual cells over time. The required enzymes and energetic demands of stomach cells are different from those of fat storage cells, skin cells, blood cells, and nerve cells. Furthermore, a digestive cell works much harder to process and break down nutrients during the time closely following a meal compared with many hours after a meal. As these cellular demands and conditions vary, so do the needed amounts and functionality of different enzymes.
Regulation of enzymes by molecules
Enzymes can be regulated in ways that either promote or reduce their activity. There are many kinds of molecules that inhibit or promote enzyme function, and various mechanisms exist for doing so. In some cases of enzyme inhibition, for example, an inhibitor molecule is similar enough to a substrate that it can bind to the active site and simply block the substrate from binding. When this happens, the enzyme is inhibited through competitive inhibition, because an inhibitor molecule competes with the substrate for active site binding. On the other hand, in noncompetitive inhibition, an inhibitor molecule binds to the enzyme in a location other than an active site and still manages to block substrate binding to the active site.
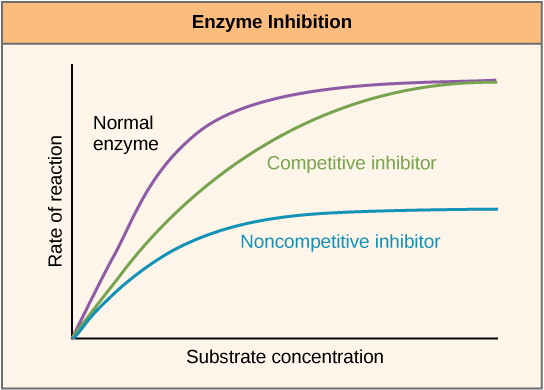
Figure 7. Competitive and noncompetitive inhibition affect the rate of reaction differently. Competitive inhibitors affect the initial rate but do not affect the maximal rate, whereas noncompetitive inhibitors affect the maximal rate.
Some inhibitor molecules bind to enzymes in a location where their binding induces a conformational change that reduces the affinity of the enzyme for its substrate. This type of inhibition is called allosteric inhibition. Most allosterically regulated enzymes are made up of more than one polypeptide, meaning that they have more than one protein subunit. When an allosteric inhibitor binds to an enzyme, all active sites on the protein subunits are changed slightly such that they bind their substrates with less efficiency. There are allosteric activators as well as inhibitors. Allosteric activators bind to locations on an enzyme away from the active site, inducing a conformational change that increases the affinity of the enzyme’s active site(s) for its substrate(s).
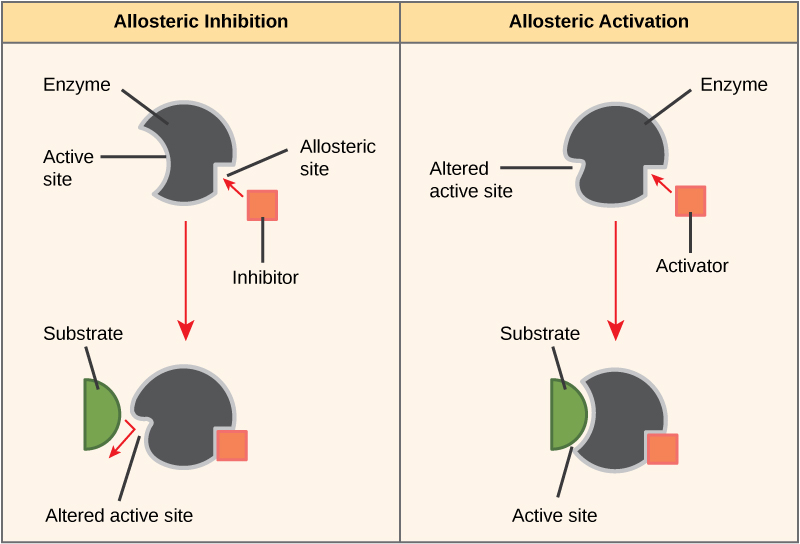
Figure 8. Allosteric inhibitors modify the active site of the enzyme so that substrate binding is reduced or prevented. In contrast, allosteric activators modify the active site of the enzyme so that the affinity for the substrate increases.
Video link
Check out this short (one-minute) video on competitive vs. noncompetitive enzymatic inhibition. Also, take a look at this video (1.2 minutes) on feedback inhibition.
Many enzymes don’t work optimally, or even at all, unless bound to other specific non-protein helper molecules, either temporarily through ionic or hydrogen bonds or permanently through stronger covalent bonds. Two types of helper molecules are cofactors and coenzymes. Binding to these molecules promotes optimal conformation and function for their respective enzymes. Cofactors are inorganic ions such as iron(II) (Fe2+) and magnesium(II) (Mg2+). One example of an enzyme that requires a metal ion as a cofactor is the enzyme that builds DNA molecules, DNA polymerase, which requires a bound zinc(II) ion (Zn2+) to function. Coenzymes are organic helper molecules, with a basic atomic structure made up of carbon and hydrogen, that are required for enzyme action. The most common sources of coenzymes are dietary vitamins. Some vitamins are precursors to coenzymes, and others act directly as coenzymes. Vitamin C is a coenzyme for multiple enzymes that take part in building the important connective tissue component, collagen. An important step in the breakdown of glucose to yield energy is catalysis by a multi-enzyme complex called pyruvate dehydrogenase. Pyruvate dehydrogenase is a complex of several enzymes that actually requires one cofactor (a magnesium ion) and five different organic coenzymes to catalyze its specific chemical reaction. Therefore, enzyme function is, in part, regulated by an abundance of various cofactors and coenzymes, which are supplied primarily by the diets of most organisms.
Enzyme compartmentalization
In eukaryotic cells, molecules such as enzymes are usually compartmentalized into different organelles. This allows for yet another level of regulation of enzyme activity. Enzymes required only for certain cellular processes can be housed separately along with their substrates, allowing for more efficient chemical reactions. Examples of this sort of enzyme regulation based on location and proximity include the enzymes involved in the latter stages of cellular respiration, which take place exclusively in the mitochondria, and the enzymes involved in the digestion of cellular debris and foreign materials, located within lysosomes.
Possible NB Discussion Point: Reversing the Effects of Caffeine
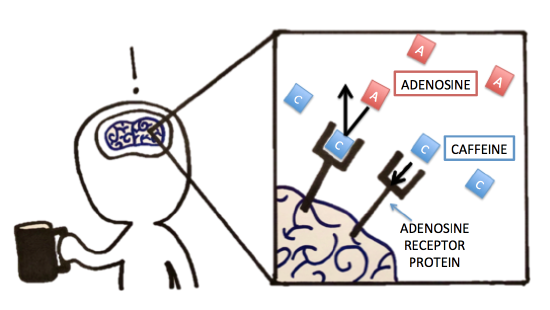
Previously, we discussed caffeine and its metabolism. Let’s now think about caffeine’s pharmacology (mode of action). Were you able to identify, compare, and contrast the molecule that caffeine had a similar structure to? Because of caffeine's structural similarity to the molecule adenosine, it is actually able to bind to the adenosine-specific receptor protein in the brain. However, because the exact lock-and-key-fit is unsatisfied, caffeine will not "activate" the adenosine receptors upon binding as adenosine would. Normally, when adenosine binds to and thereby activates its specific receptor protein in the brain, the physiological effect is increased drowsiness and muscle relaxation. It makes sense that we get tired at night because we accumulate adenosine over the day -- that's a lot of receptor activation! But back to caffeine -- when caffeine is present, it can bind to the adenosine receptor protein, thereby blocking adenosine from binding/activating the receptor. The lack of adenosine action is what leads to suppressed sleepiness and increased alertness. The inhibition seen with this receptor protein and caffeine is similar to some of the inhibition we see with enzymes. What type of inhibition would you classify this as? Follow up question: If you were hired by a company to design a solution to reverse the effect of caffeine post-ingestion, what strategies would you try to test? Explain!
Additional links
Khan Academy
The following links will take you to a series of videos on kinetics. The first link contains four videos on reaction rates, and the second link contains nine videos related to the relationship between reaction rates and concentration. These videos are supplemental and are provided to give you an outside resource to further explore enzyme kinetics.
PRACTICE POST GUIDE
General Practice
3. Why: The reaction coordinate diagram is a type of figure/abstraction commonly used to represent the energetics of a reaction. Not only is it a piece of “common language” in the sciences, once you learn how to use it, it’s a useful construct that can help you interpret a number of “how does this go” questions.
How to practice: Draw a free energy reaction coordinate diagram for both a generic endergonic and a generic exergonic reaction (label each). Make sure to label the x- and y-axes. Now label where reactants and products should go on the figure. Draw something that illustrates the free energy change for each reaction. Now write something like the following next to your figures: “reactants <—> products”.
- What do the reaction coordinate diagrams have to do with the text you just wrote?
- What is represented by the graphs that is not represented in the text?
- Which of the two reaction coordinate diagrams represents a thermodynamically spontaneous reaction?
This exercise is good practice for learning goals: “GC.39 Interpret reaction coordinate diagrams and associate changes in Gibbs enthalpy and activation energy with relative rates of reactions, equilibrium conditions, and whether a reaction is endergonic or exergonic.”
4. Why: More on reaction coordinate diagrams.
How to practice: Return to the diagram you drew in #3 and do the following. Start with the endergonic reaction. Add the term “Keq" (equilibrium constant) above the arrow in the text “reactants <—> products”. This constant is defined as: [products]/[reactants], or the ratio of the product concentration to reactant concentration at equilibrium. Do the same for the exergonic reaction. Now for each reaction write whether Keq is greater than, less than, or equal to one. If you don’t know the answers, look them up, ask TAs, or ask the instructor.
This exercise helps you practice learning objectives: “GC.38 Describe the concept of equilibrium in the context of reaction coordinate diagrams.”
5. Why: reaction coordinate diagrams continued . . .
How to practice: Use your figure from exercise #4 to make predictions about what would happen to both endergonic and exergonic reactions if (a) they were to start at equilibrium and (b) either reactants or products are added to or removed from them. We’ll return to these figures for the next lecture so don’t trash them.
This is an extension of practicing the learning objectives above and making a link to a previous learning objectives: “GC.35 Apply the concept of chemical equilibrium and the equilibrium constant to describe the progress of a chemical reaction, initially out of equilibrium, towards equilibrium, and finally at equilibrium in terms of "forward" and "reverse" reaction rates and concentrations of chemical reactants.” (From lecture 3 - review)
6. Why: We use reaction coordinates and the concepts of equilibrium in our discussion and to describe our understanding of how enzymes help reactions “go”. So here we’re going to integrate what you practiced above with the concepts of activation energies and energetic “barriers” that we use in the discussion of catalysts and enzymes.
How to practice: Using your sketchpad, draw a Gibbs enthalpy (energy) reaction coordinate diagram for an exergonic reaction and include the concept of an activation energy barrier. Label the diagram with ∆G and ∆G‡ (the double dagger notation is sometimes used to indicate the activation energy barrier). Now draw a curve for the same reaction that represents the reaction progress for a catalyzed reaction. What changed? What didn’t? Do the same for an endergonic reaction. Make sure to label reactants and products in both cases.
This is an extension of practicing the learning objectives above and making a link to a previous learning objectives: “GC.34 Interpret reaction coordinate diagrams showing either or both catalyzed and uncatalyzed reaction coordinates and identify respective activation energy barriers and relate these to the forward and reverse rates of reaction.”
7. Why: The likelihood and “strength” of molecular scale interactions and reactions in biology are associated with factors like the balance between energetically favorable and unfavorable bonds and non-covalent interactions and the entropy of a system, “before” and “after” a reaction. The interactions (bonds, etc.) and entropy (ways to spread energy in the system) contribute to ∆G of a reaction. Here I want you to start explicitly relating/thinking about how bonds and entropy contribute to ∆G by looking at a “simple” example - the dissolution of NaCl in H2O. It is intended to help you link the molecular scale happenings in the reaction to the more population-level thermodynamic quantities like ∆G, ∆H, and ∆S. Think about what kinds of bonds are lost and what kinds of new interactions are formed when NaCl dissolves in water. How do those contribute to ∆H? How does the reorganization of molecules contribute to ∆S?
How to practice: In your sketchbook, draw a free energy reaction coordinate diagram for an energy story associated with the dissociation of NaCl (table salt) in water. Recall that this reaction is spontaneous. Be sure to label the axes and the other components of the diagram (reactants, products, etc). Use each figure to tell yourself a convincing energy story for the reaction and make sure that you include the concepts of bonds forming and breaking and a change of entropy in your story.
While I don’t have an explicit learning objective, yet, for the skill practiced here (one will come), this exercise wants you to relate the skills of using reaction coordinate diagrams and the associated thermodynamics concepts together with your understanding of molecular interactions. You’re bringing together various learning objective here and will build the mental picture to help with similar ideas involving the role of enzymes.
8. Why: This practice asks you to extend on the practice you did in exercise #7 by getting you to think about the connection between the molecular events that happen in an enzyme and the associated thermodynamic quantities that describe the reaction and are represented in a reaction coordinate diagram.
How to practice: In class, we often talk about an enzyme lowering the activation energy barrier and we often draw a smooth curve that has a smaller ∆G‡ than that of the uncatalyzed reaction. However, sometimes you will see figures like the at right. Can you think of a reason why the red curve is depicted with so many bumps?
This exercise is practice for the integration of some of the learning objectives practiced above with those that describe concepts of how enzymes work. For instance: “GC.34 Interpret reaction coordinate diagrams showing either or both catalyzed and uncatalyzed reaction coordinates and identify respective activation energy barriers and relate these to the forward and reverse rates of reaction.”; & “GC.36 Describe mechanisms used by enzymes to lower the activation energy and increase rates of reaction.”
9. Why: Enzymes work to increase the rates of reactions and their activity can be modulated. Both of these processes occur because of interactions between molecules that happen at the molecular level. This encourages you to think about the link between what happens (allosteric regulation) and how it happens (the kinds of molecular interactions that might take place).

How to practice: The figure below (“For question 9”) depicts the rate of an enzyme catalyzed reaction with respect to substrate concentration. The black curve in the center is a curve showing enzyme activity when the experiment is carried out with only enzyme and substrate in the tube. The blue and red curves depict the influence of two different molecules on the rates of reactions. What is the influence of the “blue” compound? The “red” compound? What are each of these generic types of compounds called and how are they influencing the rate of reaction? (This can be found in the reading). Use a drawing of an enzyme and relevant binding sites to help explain your interpretation.
This is practice for learning objectives: “MS.18 Hypothesize how binding of small molecules to one or more binding pockets can lead to changes in protein function (i.e. competitive inhibition and/or allostery).” & “MS.17 Draw a rough sketch of an enzyme including its active site and other sites in the enzyme that might impact its function, such as an inhibitor binding site.”
10. Why: The exercises above ask you to think about and picture the molecular basis for interactions between molecules and how this can be extended to understanding/picturing how enzymes work. Here we extend this idea and ask you to imagine “What if we changed some of those interactions? How would that impact function?”
How to practice: Based on what you know about the structure of proteins, the types of bonds that hold them together, the role of amino acids in binding substrate and catalysis, try to enumerate some different ways in which specific changes in amino acid composition could influence the behavior of a protein. I suggest looking back at a table of amino acids and asking something like: “Here’s a valine. What might be a consequence of changing this to a glutamate?” You need to think about what role a valine might play in a structure and figure out how changing that might change the role. How might that role be enhanced or disrupted by a change in amino acids?
This is a revisiting of the following learning objective in the specific context of proteins: “MS. 14 Form hypotheses about how different types of changes at a molecular interface might influence binding (i.e. proteins-proteins, protein, DNA, protein-lipid, small molecule-lipid, small molecule-protein, etc.).This would include recognizing or inferring different types of chemical bonds from an analysis or knowledge of functional group interactions.”
PRACTICE EXAM QUESTIONS
Question Q6.1
Q6.1 Dissolving potassium hydroxide in water is an exothermic process (it gives up energy to the solution and surroundings) and spontaneous at room temperature. Note that exothermic and exergonic are different terms. Meanwhile, dissolving ammonium nitrate is an endothermic process and spontaneous at room temperature (the solution absorbs energy from the environment). Which of the following explains this?
- While ΔH(potassium hydroxide/water) is positive, TΔS(ammonium nitrate/water) is larger.
- While ΔH(ammonium nitrate/water) is positive, TΔS(ammonium nitrate/water) is larger.
- While ΔH(ammonium nitrate/water) is negative, TΔS(ammonium nitrate/water) is larger.
- Keq of the potassium hydroxide reaction is >1 while it is <1 for ammonium nitrate.
- Dissolving ammonium nitrate is slower than dissolving potassium hydroxide.
Question Q6.2
Q6.2 Exergonic reactions:
- are necessarily exothermic.
- are spontaneous reactions.
- create products with greater free energy than the substrates.
- All statements are true.
Question Q6.3
Q6.3 A reaction with ΔH > 0 and ΔS < 0 (and room temperature) will be:
- spontaneous
- favorable at high temperatures
- non-spontaneous
- favorable at cold temperatures
- There is insufficient data to answer the question.
Question Q6.4
Q6.4 The reaction coordinate diagram depicts the free energy profile for a reaction both catalyzed and uncatalyzed. S = substrates, P = products. The bar graphs represent hypothetical equilibrium concentrations for S and P of the same reaction. Which bar chart below best represents the equilibrium conditions of the catalyzed and uncatalyzed reaction?

Question Q6.5
Q6.5 A reaction coordinate diagram for a reaction converting S to P is shown on the right. Which combination of differences in free energy describes forward and reverse rates for the catalyzed reaction, respectively?

- 1 only
- 1 and 3
- 2 and 5
- 3 only
- 3 and 4