
3.3.1: Example - Mutations and Cystic Fibrosis
- Page ID
- 32274

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Cystic Fibrosis (CF)
Cystic fibrosis (CF) is one of many diseases that geneticists have shown to be caused by mutation of a single, well-characterized gene. Cystic fibrosis is the most common (1/2,500) life-limiting autosomal recessive disease among people of European heritage, with ~ 1 in 25 people being carriers. The frequency varies in different populations. Most of the deaths caused by CF are the result of lung disease, but many CF patients also suffer from other disorders including infertility and gastrointestinal disease. The disease is due to a mutation in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene, which was identified by Lap-chee Tsui’s group at the University of Toronto.
Epithelial tissues in some organs rely on the CFTR protein to transport ions (especially Cl-) across their cell membranes. The passage of ions through a six-sided channel is gated by another part of the CFTR protein, which binds to ATP. If there is insufficient activity of CFTR, an imbalance in ion concentration results, which disrupts the properties of the liquid layer that normally forms at the epithelial surface. In the lungs, this causes mucus to accumulate and can lead to infection. Defects in CFTR also affect pancreas, liver, intestines, and sweat glands, all of which need this ion transport. CFTR is also expressed at high levels in the salivary gland and bladder, but defects in CFTR function do not cause problems in these organs, probably because other ion transporters are able to compensate.
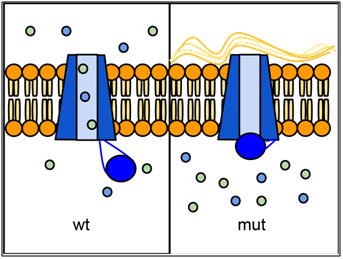
Over one thousand different mutant alleles of CFTR have been described. Any mutation that prevents CFTR from sufficiently transporting ions can lead to cystic fibrosis (CF). Worldwide, the most common CFTR allele among CF patients is called ΔF508 (delta-F508 or PHE508DEL), which is a deletion of three nucleotides that eliminates a phenylalanine (F) from position 508 of the 1480 amino acid wild-type protein. Mutation ΔF508 causes CFTR to be folded improperly in the endoplasmic reticulum (ER) and preventing CFTR from reaching the cell membrane. The allele ΔF508 accounts for approximately 70% of CF cases in North America, with ~1/25 people of European descent being carriers. The high frequency of the ΔF508 allele around the world has led to speculation that it may confer some selective advantage to heterozygotes, perhaps by reducing dehydration during cholera epidemics or by reducing susceptibility to certain pathogens that bind to epithelial membranes, although little data has been uncovered to support these hypotheses. However, recent mining of health data and genetic data from over 19,000 CFTR mutant allele heterozygotes suggests that heterozygotes exhibit an increased incidence of a variety of conditions including infertility, pancreatitis, diabetes, short stature, failure to thrive, constipation and scoliosis (Miller et al., 2020).
Query \(\PageIndex{1}\)
CFTR is also notable because it is one of the well-characterized genetic diseases for which a drug has been developed that compensates for the effects of a specific mutation. The drug, Kalydeco® (ivacaftor) made by Vertex Pharmaceuticals, was approved by the FDA and Health Canada in 2012, decades after the CFTR gene was first mapped to DNA markers (Tsui et al. 1985; Wainwright et al. 1985; White et al., 1985) and cloned (Kerem et al. 1989; Riordan et al. 1989; Rommens et al. 1989). Kalydeco is effective on only some CFTR mutations, for example G551D (i.e. where glycine is substituted by aspartic acid at position 551 of the protein; GLY551ASP). This mutation is found in less than 5% of CF patients. The G551D mutation affects the ability of ATP to bind to CFTR and open the channel it for transport. Kalydeco® compensates for the mutation by binding to CFTR and holding it in an open conformation. Kalydeco® is priced at approximately $300,000 per patient per year, but costs may be covered by insurance. In 2015 an additional drug, Orkambi® was approved. This therapy combines ivacaftor with a second drug lumacaftor, which increases the trafficking of the ΔF508 protein to the cell surface. Like Kalydeco, Orkambi costs about $300,000 per year.
Watch this video about Cystic Fibrosis Mechanism and Treatment (from HHMI BioInteractive).
Query \(\PageIndex{2}\)
Contributors and Attributions
Dr. Todd Nickle and Isabelle Barrette-Ng (Mount Royal University) The content on this page is licensed under CC SA 3.0 licensing guidelines.
References
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC 1989. Identification of the cystic fibrosis gene: genetic analysis. Science. Sep 8; 245(4922):1073-80.
Miller AC, Comellas AP, Hornick DB, Stoltz DA, Cavanaugh JE, Gerke AK, Welsh MJ, Zabner J, Polgreen PM. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proceedings of the National Academy of Sciences Jan 2020, 117 (3) 1621-1627; DOI: 10.1073/pnas.1914912117 (https://www.pnas.org/content/117/3/1621)
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. Sep 8; 245(4922):1066-73.
Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N. 1989. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. Sep 8; 245(4922):1059-65.
Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, Eiberg H, Mohr J, Kennedy D, Plavsic N, et al. 1985. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 230: 1054–1057.
Wainwright BJ, Scambler PJ, Schmidtke J, Watson EA, Law HY, Farrall M, Cooke HJ, Eiberg H, Williamson R 1985. Localization of cystic fibrosis locus to human chromosome 7cen-q22. Nature 318: 384–385.
White R, Woodward S, Leppert M, O’Connell P, Hoff M, Herbst J, Lalouel JM, Dean M, Vande Woude G 1985. A closely linked genetic marker for cystic fibrosis. Nature 318: 382–384.