
5.15: Metagenomics - Exploring the Microbial World
- Page ID
- 4763

All the genomes listed on my page Genome Sizes describe the complete genome of a single species. For bacteria and archaeons, this means that the organism was grown in pure culture to provide the DNA for sequencing. But it is now clear that the microbial world contains vast numbers of both groups that have never been grown in the laboratory and thus have escaped study. Soil, water, and the contents of our large intestine are examples of habitats that teem with unknown microorganisms.
Thanks to the recent development of sequencing machines capable of rapidly (and inexpensively) sequencing huge amounts of DNA, it is now practical to sequence the DNA extracted from complex microbial ecosystems like that found in a soil sample. Several different approaches are used, but all depend on a first step of extracting the microbial DNA from the sample (and separating it from the far more complex DNA of any eukaryotes that may be present).
Assessing Microbial Diversity
The DNA encoding the small subunit (16S) of the ribosomes of both bacteria and archaeons contain some highly conserved regions; that is, regions of identical or almost identical sequence. Using primers that target these regions, one can then produce enough material by the polymerase chain reaction PCR to sequence the entire 16S rRNA gene.
Comparing the various sequences to a database of sequences from known organisms, one can estimate how many different types of microbes are present. Because of the substantial genetic diversity found between "strains" of a single species (e.g., E. coli K-12 and E.coli O157:H7), closely-related (> 97% identity) 16S rDNA sequences are assigned to a single "phylotype" because we cannot be sure whether they belong to separate species or to two strains of the same species. In either case, the collection of 16S rDNA sequences can be arranged to form a phylogenetic tree to show the patterns of relatedness.
Cataloging the Genes in a Microbial Ecosystem
Analyzing the 16S rDNA genes in a sample tells us who is there, but, of course, is not a complete genome and tells us nothing about the other genes present in the various members of the population. This information can be gained by "shotgun" sequencing of the environmental DNA sample.
The Steps:
- Break the DNA in short fragments.
- Insert these into a vector, e.g. a plasmid capable of growing in E. coli K-12.
- Expose E. coli cells to this random mix and grow the individual bacterial cells into colonies.
- The result: a library containing millions of random DNA fragments from the original sample.
- Isolate the plasmids and sequence them. Sequence "reads" average around 100 nucleotides — far shorter than a gene but often enough to move on to the next step.
- Use a powerful computer to attempt to assemble the fragments into a linear sequence of DNA. The computer looks for identical stretches of nucleotides in different fragments and uses the overlap to assemble them into a "contig".
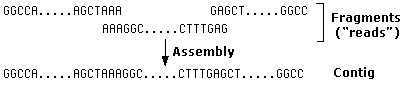
- Look (have the computer look) for open reading frames (ORFs) of protein-encoding genes.
- Compare the ORFs with those of known microbes already in databases to see if a function can be deduced.
The sheer diversity of organisms in most microbial ecosystems makes it virtually impossible to find enough contigs to assemble a complete genome for any one organism like those listed in Genome Sizes. What you get instead is a window into the many kinds of genes present in one inhabitant or another of that ecosystem. For example, you may discover genes that encode proteins able to degrade environmental pollutants or genes able to synthesize a new antibiotic.
Finding New Functions in Microbial Populations
Another way of exploiting metagenomics is to look for new functions in the host (e.g. E. coli) if it can express the new gene with which it was transformed. For example, screening the library of E. coli clones for the ability to resist an antibiotic can reveal genes involved in antibiotic resistance — a worrisome development in recent years.
Some Applications of Metagenomics
- The Sargasso Sea: Metagenomic analysis of the DNA extracted from sea water in the Sargasso Sea revealed the presence of over a thousand different 16S rDNA genes (and thus approximately that number of different species) and over a million protein-encoding genes.
- The Human Colon: 0.3 g fecal samples from two healthy humans produced 78 million base pairs of sequence. Each subject produced some 25 thousand open reading frames (ORFs) of which about half could be recognized as already-known bacterial or archaeal genes. Included were genes encoding enzymes for the synthesis of vitamins (e.g., vitamin B1), amino acids, and enzymes for the digestion of complex polysaccharides in our diet which would otherwise be indigestible. Perhaps as much as 10% of the energy we extract from our food is made available to us by the activity of these microorganisms.
- Acid Mine Drainage: Metagenomic analysis of the acidic water (pH ~0.5) flowing from an abandoned metal mine in California revealed a much simpler ecosystem than those described above: only 3 species of bacteria and 2 of archaea. With such limited diversity, it was possible to assemble almost-complete genomes for two of these organisms.
- A South African Gold Mine: Simpler still was the ecosystem found in water 2.8 km (1.7 miles) down in a gold mine. Only one organism turned up: an autotrophic bacterium capable of extracting energy from inorganic substances in its environment and synthesizing all the molecules needed for its life from them.