
2.8: Intro
- Page ID
- 13608
DNA and RNA are both nucleic acids, which are the polymeric acids isolated from the nucleus of cells. DNA and RNA can be represented as simple strings of letters, where each letter corresponds to a particular nucleotide, the monomeric component of the nucleic acid polymers. Although this conveys almost all the information content of the nucleic acids, it does not tell you anything about the underlying chemical structures. This chapter will be review the evidence that nucleic acids are the genetic material, and then exploring the chemical structure of nucleic acids.
Southern blot-hybridizations
After separation by electrophoresis, DNA fragments are transferred to a membrane (nylon or nitrocellulose) and immobilized; this replica of the DNA pattern in the gel is called a "blot." A specific labeled probe is hybridized to the blot to detect related sequences. After nonspecifically bound probe is washed away, the specific hybrids are detected by autoradiography of the blot.

Restriction sites can be used as genetic markers. One can identify restriction fragment length polymorphisms (RFLPs) that are linked to a particular locus. This can be be used to
- Develop a diagnostic test for a disease locus (e.g. sickle cell disease)
- Help isolate the gene.
- DNA fingerprinting for highly variable loci.
Sizes of DNAs and chromosomes, and methods to resolve them
The next figure presents views of chromosomes and DNA segments on four different, expanding scales. The top level compares the sizes of intact chromosomes from four of the organisms we will be discussing in this course. The scale on yeast chromosome III is then expanded so that it can be compared to some of the viral and plasmid genomes that are in common use. Next, a higher resolution view of the plasmid pBR322 is given, and finally the highest resolution that we are usually concerned with, i.e. the nucleotide sequence.

Determining the sequence of DNA and RNA
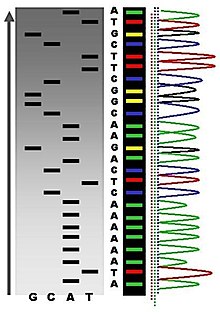
The basic approach is to generate a nested set of DNA fragmentsthat start a common site and end in either A, G ,C or T. These sets of (labeled) DNA fragments are separated on a denaturing polyacrylamide gel that has a resolution of 1 bp. The resulting pattern allows the sequence to be read. Base-specific chemical modification and degradation, developed by Maxam and Gilbert, was a widely used approach. Nucleotide-specific cleavage of RNA by a set of Rnases can be used to sequence RNA. We will focus on the most common method of sequencing DNA, that of nucleotide-specific chain termination.
The dideoxynucleotide chain termination methodwas developed in the laboratory of Fred Sanger at Cambridge. A 2’, 3’ dideoxynucleotide can be incorporated into DNA, as directed by the template strand. However, the missing 3’-OH precludes further polymerization. Hence the newly synthesized chain of nucleotides ends at base-specific, chain terminating dideoxynucleotide. Reactions are run such that all the products end in a G, a C, an A, or aT, but they all begin at the same place. This generates a nested set of products whose length is a measure of the position of all G’s in a target sequence, or all C’s, etc. Thus one can deduce that the target sequence is complementary to, e.g. G at position 1, T at positon 2, C at positions 3 and 4, etc. for hundreds of nucleotides per run.
In more detail, a specific primer is annealed to the template, upstream from the region to be sequenced. DNA polymerase will catalyze the synthesis of new DNA from the 3' end of that primer (elongation). The primer therefore generates a common end to all the product fragments. (This is the basis for the nested set in this approach).
The synthesized DNA is labeled with either a radioactive nucleotide, such as [a35S]deoxy‑thio‑ATP, or a fluorescent dye, often attached to the primer.
A base-specific chain‑terminator is included in each of four reactions:
- 2',3' dideoxyGTP in the "G" reaction.
- 2',3' dideoxyATP in the "A" reaction.
- 2',3' dideoxyTTP in the "T" reaction.
- 2',3' dideoxyCTP in the "C" reaction.
The DNA polymerase will elongate from each annealed primer until it incorporates a 2', 3' dideoxynucleotide. No additional nucleotides can be added to this product, since it has no 3' OH, thus it is a chain-terminator. This termination occurs only at G residues (complementary to C's in the template) in the "G" reaction, only at A residues in the "A" reaction, etc. Thus the products of each reaction comprise a nested set of fragments, with the specific primer at the 5' end and the base-specific chain terminator at the 3' end. The products are resolved on a sequencing gel, exposed to X-ray film and the sequence read, as in Figure 2.30.

The dideoxynucleotide chain-termination approach is the method used in automated sequenators. Different color fluorescent dyes (usually attached to the primer) are included in each base-specific reaction. Therefore the products of all four can be run in 1 lane of the resolving gel, allowing >20 sequencing sets to be analyzed at one time. A laser scans continuously along one zone of the gel, and records when a (e.g.) red, green, blue or yellow fluoresence is detected in each lane, meaning that the primer extended to a (e.g.) A, G, C or T is passing through the detection zone. These data are automatically processed, and a readout is generated with the peaks for each fluorescent dye as function of time of the gel running and the deduced sequence. An example of the output is shown below in black-and-white; the original output is in color (a different color for each nucleotide). Manual editing of the deduced sequence can be done based on the raw data, but in large scale sequencing projects, each region is determined about 8 different times and other software is used to determine the most frequently ocurring nucleotide at each position.
The capacity of automated sequencing machines is extraordinary. New machines using capillary gel electrophoresis are used to generated millions of nucleotides per day in the major sequencing centers. This technology allows large, complex genomes to be sequenced rapidly, as discussed in Chapter 4.
Supercoiling of Topologically Constrained DNA
Topologically closed DNA can be circular (covalently closed circles) or loops that are constrained at the base. The coiling (or wrapping) of duplex DNA around its own axis is called supercoiling (Figure 2.32 middle).
- Negative supercoils twist the DNA about its axis in the opposite direction from the clockwise turns of the right-handed (R-H) double helix.
- Negatively supercoiled DNA is underwound (and thus favors unwinding of duplex).
- Negatively supercoiled DNA has R-H supercoil turns (Figure 2.32).
- Positive supercoils twist the DNA in the same direction as the turns of the R-H double helix.
- Positively supercoiled DNA is overwound (helix is wound more tightly).
- Positively supercoiled DNA has L-H supercoil turns.
The clockwise turns of R-H double helix (A or B form) generate a positive Twist (T); see Figure 2.32 left. The couterclockwise (ccw) turns of L-H helix (Z ) generate a negative T.
T= Twisting number
- For B form DNA, it is + (# bp/10 bp per twist)
- For A form DNA, it is + (# bp/11 bp per twist)
- For Z DNA, it is - (# bp/12 bp per twist)
W= Writhing Numberis the turning of the axis of the DNA duplex in space
- Relaxed molecule W=0
- Negative supercoils, W is negative
- Positive supercoils, W is positive
L= Linking number= total number of times one strand of the double helix (of a closed molecule) encircles (or links) the other.
\[L = W + T\]
- L cannot change unless one or both strands are broken and reformed.
- A change in the linking number, DL, is partitioned between T and W (Figure 2.32 right). Thus:
\[DL=DW+DT\]
if \(DL = 0\), \(DW=-DT\)

Ethidium Bromide intercalates in DNA, and untwists (or unwinds) the duplex by -27° per molecule of ethidium bromide intercalated. Thus intercalation of 14 molecules of ethidium bromide will untwist the duplex by 378o, i.e. slightly more than one full twist (which would be 360°). For this process of intercalation, DL=0, since no covalent bonds in the DNA are broken or reformed. The change in twist, DT, is negative, and thus DW is positive. Thus intercalation of ethidium bromide can relax a negatively supercoiled circle, and further intercalation will make the DNA positively supercoiled (Figure 2.33).

It is useful to have an expression for supercoiling that is independent of length. The superhelical density is simply the number of superhelical (S.H.) turns per turn (or twist) of double helix.
\[\text{Superhelical density} = s= \dfrac{W}{T}\]
This is -0.05 for natural bacterial DNA. i.e., in bacterial DNA, there is 1 negative S.H. turn per 200 bp (calculated from 1 negative S.H. turn per 20 twists = 1 negative S.H. turn per 200 bp)
Negative supercoiled DNA has energy stored that favors unwinding, or a transition from B-form to Z DNA.
For s = -0.05, \(\Delta G=-9 Kcal/mole\), which favoring unwinding
Thus negative supercoiling could favor initiation of transcription and initiation of replication.
Topoisomerases
Topoisomerases catalyze a change in the linking number of DNA.
- Topo I = nicking-closing enzyme, can relax positive or negative supercoiled DNA, makes a transient break in 1 strand. E. coli Topo I specifically relaxes negatively supercoiled DNA. Calf thymus Topo I works on both negatively and positively supercoiled DNA.
- Topo II = gyrase: uses the energy of ATP hydrolysis to introduce negative supercoils. Its mechanism of action is to make a transient double strand break, pass a duplex DNA through the break, and then re-seal the break.
Measuring a change in linking number
One can measure a change in linking number (DL) by sedimentation, electrophoresis, or electron microscopy, as illustrated in Figure 2.34.

