
13.3: Calculating Likelihoods for State-Dependent Diversification Models
- Page ID
- 21658

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)To calculate likelihoods for state-dependent diversification models we use a pruning algorithm with calculations that progress back through the tree from the tips to the root. We will follow the description of this algorithm in Maddison et al. (2007). We have already used this approach to derive likelihoods for constant rate birth-death models on trees (Chapter 12), and this derivation is similar.
We consider a phylogenetic tree with data on the character states at the tips. For the purposes of this example, we will assume that the tree is complete and correct – we are not missing any species, and there is no phylogenetic uncertainty. We will come back to these two assumptions later in the chapter.
We need to obtain the probability of obtaining the data given the model (the likelihood). As we have seen before, we will calculate that likelihood going backwards in time using a pruning algorithm (Maddison et al. 2007). The key principle, again, is that if we know the probabilities at some point in time on the tree, we can calculate those probabilities at some time point immediately before. By applying this method successively, we can move back towards the root of the tree. We move backwards along each branch in the tree, merging these calculations at nodes. When we get to the root, we have the probability of the data given the model and the entire tree – that is, we have the likelihood.
The other essential piece is that we have a starting point. When we start at the tips of the tree, we assume that our character states are fixed and known. We use the fact that we know all of the species and their character states at the present day as our starting point, and move backwards from there (Maddison et al. 2007). For example, for a species with character state 0, the likelihood for state 0 is 1, and for state 1 is zero. In other words, at the tips of the tree we can start our calculations with a probability of 1 for the state that matches the tip state, and 0 otherwise.
This discussion also highlights the fact that incorporating uncertainty and/or variation in tip states for these algorithms is not computationally difficult – we just need to start from a different point at the tips. For example, if we are completely unsure about the tip state for a certain taxa, we can begin with likelihoods of 0.5 for starting in state 0 and 0.5 for starting in state 1. However, such calculations are not commonly implemented in comparative methods software.
We now need to consider the change in the likelihood as we step backwards through time in the tree (Maddison et al. 2007). We will consider some very small time interval Δt, and later use differential equations to find out what happens in the limit as this interval goes to zero (Figure 13.2). Since we will eventually take the limit as Δt → 0, we can assume that the time interval is so small that, at most, one event (speciation, extinction, or character change) has happened in that interval, but never more than one. We will calculate the probability of the observed data given that the character is in each state at time t, again measuring time backwards from the present day. In other words, we are considering the probability of the observed data if, at time t, the character state were in state 0 [p0(t)] or state 1 [p1(t)]. For now, we can assume we know these probabilities, and try to calculate updated probabilities at some earlier time t + Δt: p0(t + Δt) and p1(t + Δt).
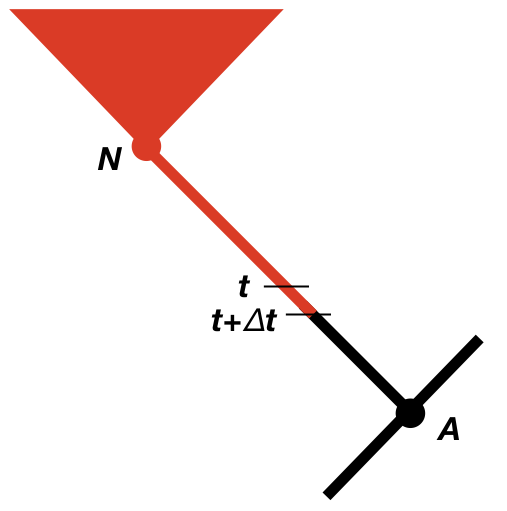
To calculate p0(t + Δt) and p1(t + Δt), we consider all of the possible things that could happen in a time interval Δt along a branch in a phylogenetic tree that are compatible with our dataset (Figure 13.2; Maddison et al. 2007). First, nothing at all could have happened; second, our character state could have changed; and third, there could have been a speciation event. This last event might seem incorrect, as we are only considering changes along branches in the tree and not at nodes. If we did not reconstruct any speciation events at some point along a branch, then how could one have taken place? The answer is that a speciation event could have occurred but all taxa descended from that branch have since gone extinct. We must also consider the possibility that either the right or the left lineage went extinct following the speciation event; that is why the speciation event probabilities appear twice in Figure 13.2 (Maddison et al. 2007).
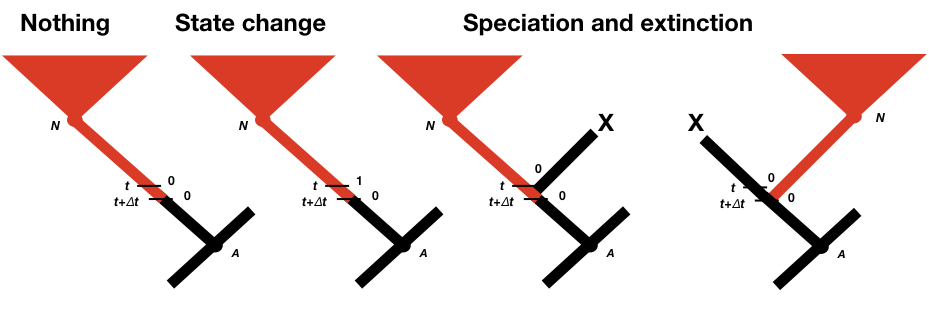
We can write an equation for these updated probabilities. We will consider the probability that the character is in state 0 at time t + Δt; the equation for state 1 is similar (Maddison et al. 2007).
\[ \begin{aligned} p_0 (t+\Delta t)=(1-\mu_0 )\Delta t \cdot [(1-q_{01} \Delta t)(1-\lambda_0 \Delta t) p_0 (t)+q_{01} \Delta t(1-\lambda_0 \Delta t) \\ p_1 (t)+2 \cdot (1-q_{01} \Delta t) \lambda_0 \Delta t \cdot E_0 (t) p_0 (t)] \end{aligned} \label{13.4}\]
We can multiply through and simplify. We will also drop any terms that include [Δt]2, which become vanishingly small as Δt decreases. Doing that, we obtain (Maddison et al. 2007):
\[p_0(t + Δt)=[1 − (λ_0 + μ_0 + q_{01})Δt]p_0(t)+(q_{01}Δt)p_1(t)+2(λ_0Δt)E_0(t)p_0(t) \label{13.5}\]
Similarly,
\[p_1(t + Δt)=[1 − (λ_1 + μ_1 + q_{10})Δt]p_1(t)+(q_{10}Δt)p_0(t)+2(λ_1Δt)E_1(t)p_1(t) \label{13.6}\]
We can then find the instantaneous rate of change for these two equations by solving for p1(t + Δt)/[Δt], then taking the limit as Δt → 0. This gives (Maddison et al. 2007):
\[ \frac{dp_0}{dt} = -(\lambda_0 + \mu_0 + q_{01}) p_0(t) + q{01}p_1(t) + 2 \lambda_0 E_0(t) p_0(t) \label{13.7}\]
and:
\[ \frac{dp_1}{dt} = -(\lambda_1 + \mu_1 + q_{10}) p_1(t) + q{10}p_1(t) + 2 \lambda_1 E_1(t) p_1(t) \label{13.8}\]
We also need to consider E0(t) and E1(t). These represent the probability that a lineage with state 0 or 1, respectively, and alive at time t will go extinct before the present day. Neglecting the derivation of these formulas, which can be found in Maddison et al. (2007) and is closely related to similar terms in Chapters 11 and 12, we have:
\[ \frac{dE_0}{dt} = \mu_0-(\lambda_0+\mu_0+q_{01} ) E_0 (t)+q_{01} E_1 (t)+\lambda_0 [E_0 (t)]^2 \label{13.9} \]
and:
\[ \frac{dE_1}{dt} = \mu_1-(\lambda_1+\mu_1+q_{10} ) E_1 (t)+q_{10} E_0 (t)+\lambda_1 [E_1 (t)]^2 \label{13.10}\]
Along a single branch in a tree, we can sum together many such small time intervals. But what happens when we get to a node? Well, if we consider the time interval that contains the node, then we already know what happened – a speciation event. We also know that the two daughters immediately after the speciation event were identical in their traits (this is an assumption of the model). So we can calculate the likelihood for their ancestor for each state as the product of the likelihoods of the two daughter branches coming into that node and the speciation rate (Maddison et al. 2007). In this way, we merge our likelihood calculations along each branch when we get to nodes in the tree.
When we get to the root of the tree, we are almost done – but not quite! We have partial likelihood calculations for each character state – so we know, for example, the likelihood of the data if we had started with a root state of 0, and also if we had started at 1. To merge these we need to use probabilities of each character state at the root of the tree (Maddison et al. 2007). For example, if we do not know the root state from any outside information, we might consider root probabilities for each state to be equal, 0.5 for state 0 and 0.5 for state 1. We then multiply the likelihood associated with each state with the root probability for that state. Finally, we add these likelihoods together to obtain the full likelihood of the data given the model.
The question of which root probabilities to use for this calculation has been discussed in the literature, and does matter in some applications. Aside from equal probabilities of each state, other options include using outside information to inform prior probabilities on each state (e.g. Hagey et al. 2017), finding the calculated equilibrium frequency of each state under the model (Maddison et al. 2007), or weighting each root state by its likelihood of generating the data, effectively treating the root as a nuisance parameter (FitzJohn et al. 2009).
I have described the situation where we have two character states, but this method generalizes well to multi-state characters (the MuSSE method; FitzJohn 2012). We can describe the evolution of the character in the same way as described for multi-state discrete characters in chapter 9. We then can assign unique diversification rate parameters to each of the k character states: λ0, λ1, …, λk and μ0, μ1, …, μk (FitzJohn 2012). It is worth keeping in mind, though, that it is not too hard to construct a model where parameters are not identifiable and model fitting and estimation become very difficult.