
5.4: Whole-Genome Alignment
- Page ID
- 40938

Once we have access to whole-genome sequences for several different species, we can attempt to align them in order to infer the path that evolution took to differentiate these species. In this section we discuss some of the methods for performing whole-genome alignments between multiple species.
Global, local, and ’glocal’ alignment
The Needleman-Wunsch algorithm discussed in chapter 2 is the best way to generate an optimal alignment between two or more genome sequences of limited size. At the level of whole genomes, however, the O(n2) time bound is impractical. Furthermore, in order to find an optimal alignment between k different species, the time for the Needleman-Wunsch algorithm is extended to O(nk). For genomes that are millions of bases long, this run time is prohibitive (Figure 5.15).
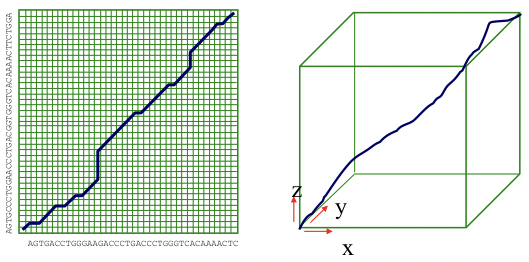
Figure 5.15: The Needleman-Wunsch algorithm for alignments of 2 and 3 genomes
One alternative is to use an efficient local alignment tool such as BLAST to find all of the local alignments, and then chain them together along the diagonal to form global alignments. This approach can save a significant amount of time, since the process of finding local alignments is very efficient, and then we only need to perform the time-consuming Needleman-Wunsch algorithm in the small rectangles between local alignments (Figure 5.16).
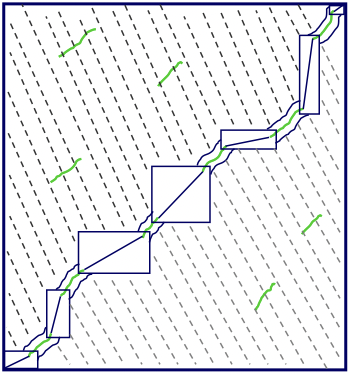
Figure 5.16: We can save time when performing a global alignment by first finding all the local alignments and then chaining them together along the diagonal with restricted dynamic programming.
Another novel approach to whole genome alignment is to extend the local alignment search to include inversions, duplications and translocations. Then we can chain these elements together using the least-cost transformations between sequences. This approach is commonly called glocal alignment, since it seeks to combine the best of local and global alignment to create the most accurate picture of how genomes evolve over time (Figure 5.17).
Lagan: Chaining local alignments
LAGAN is a popular software toolkit that incorporates many of the above ideas and can be used for local, global, glocal, and multiple alignments between species.
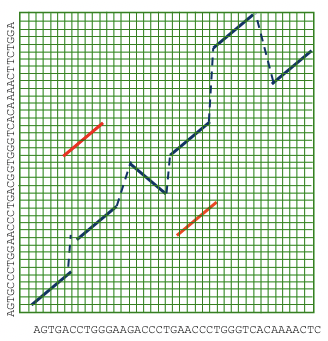
Figure 5.17: Glocal alignment allows for the possibility of duplications, inversion, and translocations.
The regular LAGAN algorithm consists of finding local alignments, chaining local alignments along the diagonal, and then performing restricted dynamic programming to find the optimal path between local alignments.
Multi-LAGAN uses the same approach as regular LAGAN but generalizes it to multiple species alignment. In this algorithm, the user must provide a set of genomes and a corresponding phylogenetic tree. Multi- LAGAN performs pairwise alignment guided by the phylogenetic tree. It first compares highly related species, and then iteratively compares more and more distant species.
Shuffle-LAGAN is a glocal alignment tool that finds local alignments, builds a rough homology map, and then globally aligns each of the consistent parts (Figure 5.18). In order to build a homology map, the algorithm chooses the maximum scoring subset of local alignments based on certain gap and transformation penalties, which form a non-decreasing chain in at least one of the two sequences. Unlike regular LAGAN, all possible local alignment sequences are considered as steps in the glocal alignment, since they could represent translocations, inversions and inverted translocations as well as regular untransformed sequences. Once the rough homology map has been built, the algorithm breaks the homologous regions into chunks of local alignments that are roughly along the same continuous path. Finally, the LAGAN algorithm is applied to each chunk to link the local alignments using restricted dynamic programming.
By running Shuffle-LAGAN or other glocal alignment tools, we can discover inversions, translocations, and other homologous relations between different species. By mapping the connections between these rear- rangements, we can gain insight into how each species evolved from the common ancestor (Figure 5.19).