
5.5: Gene-based region alignment
- Page ID
- 40939

An alternative way for aligning multiple genomes anchors genomic segments based on the genes that they contain, and uses the correspondence of genes to resolve corresponding regions in each pair of species. A nucleotide-level alignment is then constructed based on previously-described methods in each multiply- conserved region.
Because not all regions have one-to-one correspondence and the sequence is not static, this is more difficult: genes undergo divergence, duplication, and losses and whole genomes undergo rearrangements. To help overcome these challenges, researchers look at the amino-acid similarity of gene pairs across genomes and the locations of genes within each genome.
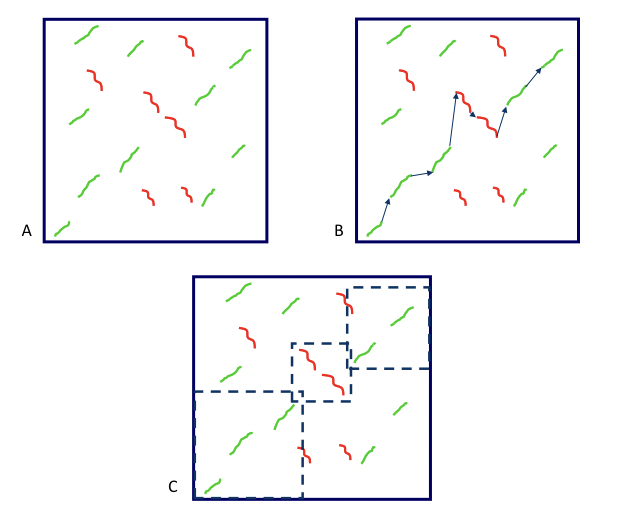
Figure 5.18: The steps to run the SLAGAN algorithm are A. Find all the local alignments, B. Build a rough homology map, and C. globally align the consistent parts using the regular LAGAN algorithm
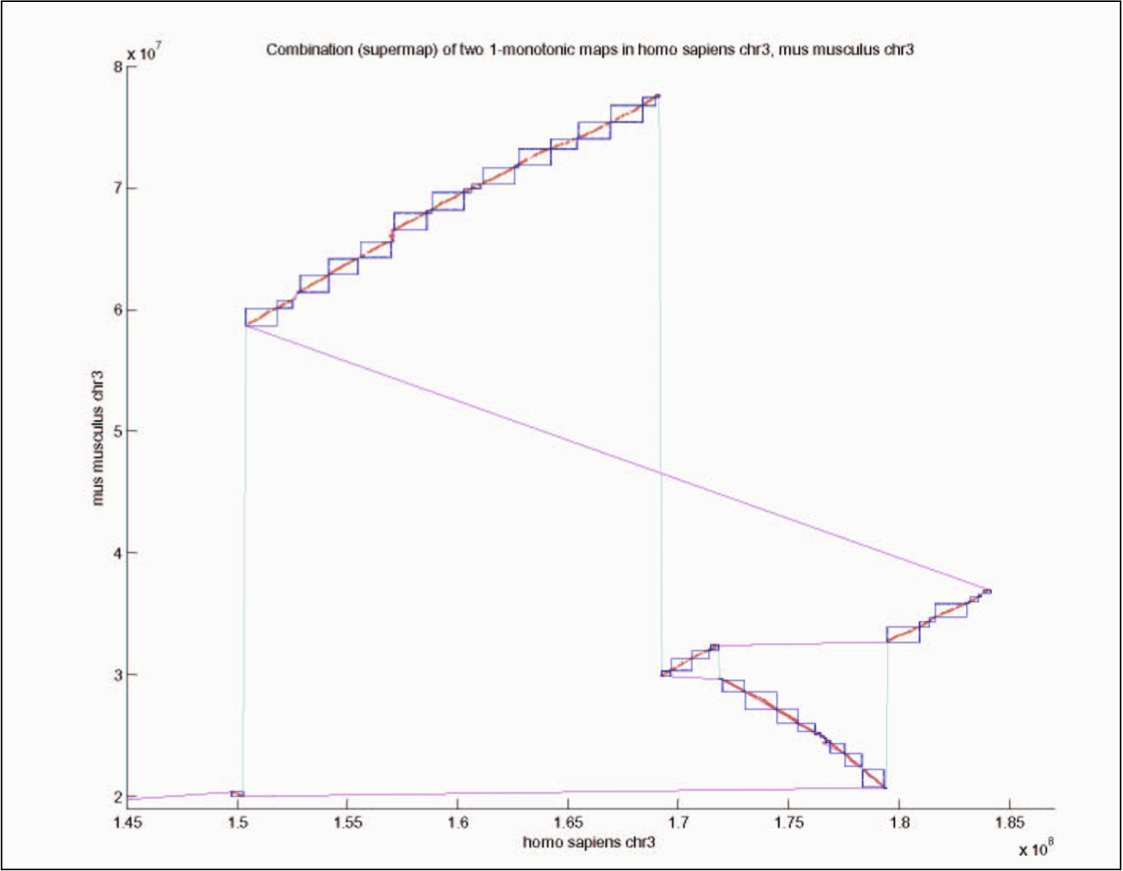
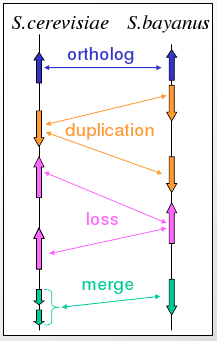
Figure 5.20: Graph of S. cerevisae and S. bayanus gene correspondence.
Gene correspondence can be represented by a weighted bipartite graph with nodes representing genes with coordinates and edges representing weighted sequence similarity (Figure 5.20). Orthologous relationships are one-to-one matches and paralogous relationships are one-to-many or many-to-many matches. The graph is first simplified by eliminating spurious edges and then edges are selected based on available information such as blocks of conserved gene order and protein sequence similarity.
The Best Unambiguous Subgroups (BUS) algorithm can then be used to resolve the correspondence of genes and regions. BUS extends the concept of best-bidirectional hits and uses iterative refinement with an increasing relative threshold. It uses the complete bipartite graph connectivity with integrated amino acid similarity and gene order information.
Did You Know?
ThA bipartite graph is a graph whose vertices can be split into two disjoint sets U and V such that every edge connects a vertex in U to a vertex in V.

Figure 5.21: Illustration of gene correspondence for S.cerevisiae Chromosome VI (250-300bp).
In the example of a correctly resolved gene correspondence of S.cerevisiae with three other related species, more than 90% of the genes had a one-to-one correspondence and regions and protein families of rapid change were identified.