
4.4: Diversity of evolutionary signatures- An Overview of Selection Patterns
- Page ID
- 40930

Independently of the substitution rate, we may also consider the pattern of substitutions in a particular nucleotide subsequence. Consider a sequence of nucleotides which encodes a protein. Due to tRNA wobble, a mutation in the third nucleotide of a codon is less likely to affect the final protein than a mutation in the other positions. Hence we expect to see a pattern of increased substitutions on the third position when looking at protein–coding subsequences of the genome. This is indeed verified experimentally, as shown in Figure 4.11.

Figure 4.11: Different mutation patterns in protein–coding and non–protein–coding regions. Asterisks in- dicate that the nucleotide was conserved across all species. Note that within the protein–coding exon, nucelotides 1 and 2 of each codon tend to be conserved, while codon 3 is allowed to vary more, which is consistent with the phenomenon of wobble.
FAQ
Q: In Figure 4.11, we also see nucleotide substitutions in groups of three or sixes. Why is this the case?
A: Insertions and deletions in groups of threes and sixes also contribute to preserving the reading frame. If all the nucleotides are deleted in one codon, the rest of the codons are unaffected during amino acid translation. However, if we delete a number of nucleotides that is not a multiple of three (i.e. we only delete part of some codon), then the translation of the rest of the codons become nonsensical since the reading frame has been shifted.
In Figure 4.12, we can see one more feature of protein-coding genes. The boundaries of conservation are very distinct and they lie near splice sites. Periodic mutations (in multiples of three) begin to occur after the splice site boundary.
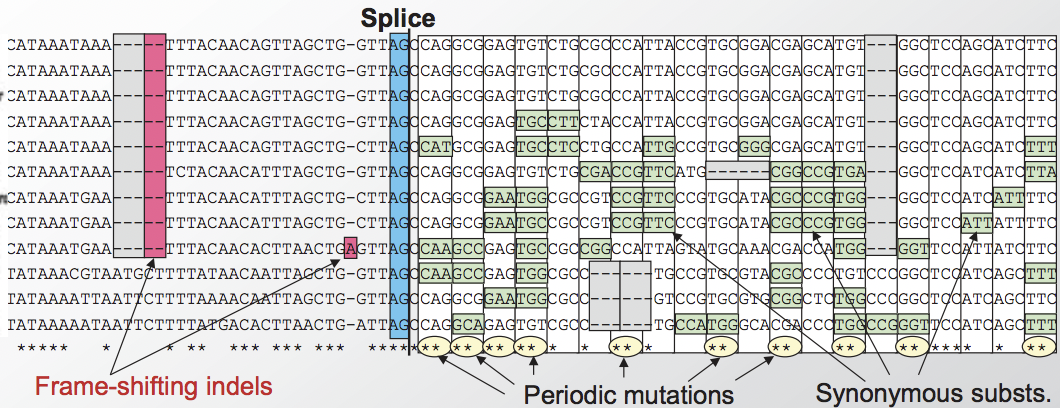
Figure 4.12: In addition to reading frame conservation and substitutions every third nucleotide, we also see sharp conservation boundaries that pinpoint splice sites.
As we can see with detecting protein-coding genes, it is not only important to consider the substitution rate but also the pattern of substitutions. By observing how regions are conserved, instead of just looking at the amount of conservation, we can observe ‘evolutionary signatures’ of conservation for different functional elements.
Selective Pressures On Different Functional Elements
Different functional elements have different selective pressures (due to their structure and other characteristics); some changes (insertions, deletions, or mutations) that can be extremely harmful to one functional element may be innocuous to another. By figuring out what the “signatures” are for different elements, we can more accurately annotate a region by observing the patterns of conservation it shows.
Such a pattern is called an evolutionary signature: a pattern of change that is tolerated within elements that still preserve their function. An evolutionary signature is different from the degree of conservation in that you tolerate mutation, but only specific types of mutations in specific places. Evolutionary signatures arise because evolution and natural selection are acting on different levels in certain functional elements. For instance, in a protein-coding gene evolution is acting on the level of amino acids, and so natural selection will not filter out nucleotide changes which do not affect the amino acid sequence. Whereas a structural RNA will have pressure to preserve nucleotide pairs, but not necessarily individual nucleotides.
Importantly, the pattern of conservation has a distinct phylogenetic structure. More similar species (mammals) group together with shared conserved domains that fish lack, suggesting a mammalian specific innovation, perhaps for regulatory elements not shared by fish. Meanwhile, some features are globally conserved, suggesting a universal significance, such as protein coding. Initial approximate annotation of protein coding regions in the human genome was possible using the simple heuristic that if it was conserved from human to fish it likely served as a protein coding region.
An interesting idea for a final project would be to map divergences in the multiple alignment and call these events “births” of new coding elements. By focusing on a particular element (say microRNAs) one could identify periods of innovation and isolate portions of a phylogenetic tree enriched for certain classes of these elements.
The rest of the chapter will focus on quantifying the degree to which a sequence follows a given pattern. Kellis compared the process of evolution to exploring a fitness landscape, with the fitness score of a particular sequence constrained by the function it encodes. For example, protein coding genes are constrained by selection on the translated product, so synonymous substitutions in the third base pair of a codon are tolerated.
Below is a summary of the expected patterns followed by various functional elements:
- Protein–coding genes exhibit particular frequencies of codon substitution as well as reading frame conservation. This makes sense because the significance of the genes is the proteins they code for; therefore, changes that result in the same or similar amino acids can be easily tolerated, while a tiny change that drastically changes the resulting protein can be considered disastrous. In addition to the error correction of the mismatch repair system and DNA polymerase itself, the redundancy of the genetic code provides an additional level of intrinsic error correction/tolerance.
- Structural RNA is selected based on the secondary sequence of the transcribed RNA, and thus requires compensatory changes. For example, some RNA has a secondary stem–loop structure such that sections of its sequence bind to other sections of its sequence in its “stem”, as shown in figure 4.13.
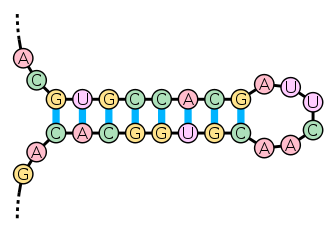
Figure 4.13: RNA with secondary stem–loop structure
Imagine that a nucleotide (A) and its partner (T) bind to each other in the stem, and then (A) mutates to a (C). This would ruin the secondary structure of the RNA. To correct this, either the (C) would mutate back to an (A), or the (T) would mutate to a (G). Then the (C)-(G) pair would maintain the secondary structure. This is called a compensatory mutation. Therefore, in RNA structures, the amount of change to the secondary structure (e.g. stem–loop) is more important than the amount of change in the primary structure (just the sequence). Understanding the effects of changes in RNA structure requires knowledge of the secondary structure. The likely secondary structure of an RNA can be determined by modeling the stability of many possible conformations and choosing the most likely conformation.
- MicroRNA is a molecule that is ejected from the nucleus into the cytoplasm. Their characteristic trait is that they also have the hairpin (stem–loop) structure illustrated in Figure 4.13, but a section of the stem is complementary to a portion of mRNA.
- When microRNA binds its complementary sequence to the respective portion of mRNA, it degrades the mRNA. This means that it is a post–transcriptional regulator, since it’s being used to limit the production of a protein (translation) after transcription. MicroRNA is conserved differently than structural RNA. Due to its binding to an mRNA target, the region of binding is much more conserved to maintain target specificity.
- Finally, regulatory motifs are conserved in sequence (to bind particular interacting protein partners) but not necessarily in location. Regulatory motifs can move around since they only need to recruit a factor to a particular region. Small changes (insertions and deletions) that preserve the consensus of the motif are tolerated, as are changes upstream and downstream that move the location of the motif.
When trying to understand the role of conservation in functional class prediction, an important question is how much of observed conservation can be explained by known patterns. Even after accounting for “random” conservation, roughly 60% of non–random conservation in the fly genome was not accounted for — that is, we couldn’t identify it as a protein–coding gene, RNA, microRNA, or regulatory motif. The fact that they remain conserved however suggests a functional role. That so much conserved sequence remains poorly understood underscores that many exciting questions remain to be answered. One final project for 6.047 in the past was using clustering (unsupervised learning) to account for the other conservation. It developed into an M.Eng project, and some clusters were identified, but the function of these clusters was, and is, still unclear. It’s an open problem!