
2.3: Problem Formulations
- Page ID
- 40913

In this section, we introduce a simple problem, analyze it, and iteratively increase its complexity until it closely resembles the sequence alignment problem. This section should be viewed as a warm-up for Section 2.5 on the Needleman-Wunsch algorithm.
Formulation 1: Longest Common Substring
As a first attempt, suppose we treat the nucleotide sequences as strings over the alphabet A, C, G, and T. Given two such strings, S1 and S2, we might try to align them by finding the longest common substring between them. In particular, these substrings cannot have gaps in them.
As an example, if S1 = ACGTCATCA and S2 = TAGTGTCA (refer to Figure 2.4), the longest common substring between them is GTCA. So in this formulation, we could align S1 and S2 along their longest common substring, GTCA, to get the most matches. A simple algorithm would be to try aligning S1 with different offsets of S2 and keeping track of the longest substring match found thus far. Note that this algorithm is quadratic in the length of the shortest sequence, which is slower than we would prefer for such a simple problem.
Formulation 2: Longest Common Subsequence (LCS)
Another formulation is to allow gaps in our subsequences and not just limit ourselves to substrings with no gaps. Given a sequence , \( \mathrm{X}=\left(x_{1}, \ldots, x_{m}\right) \) we formally define \( Z=\left(z_{1}, \ldots, z_{k}\right) \) to be a subsequence of X if there exists a strictly increasing sequence \( i_{1}<i_{2}<\ldots<i_{k} \) of indices of X such that for all j, \( x_{i_{j}}=z_{j} \) (CLRS 350-1).
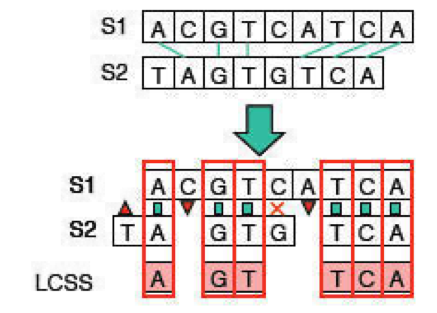
In the longest common subsequence (LCS) problem, we’re given two sequences X and Y and we want to find the maximum-length common subsequence Z. Consider the example of sequences S1 = ACGTCATCA and S2 = TAGTGTCA (refer to Figure 2.5). The longest common subsequence is AGTTCA, a longer match than just the longest common substring.
Formulation 3: Sequence Alignment as Edit Distance
Formulation
The previous LCS formulation is close to the full sequence alignment problem, but so far we have not specified any cost functions that can differentiate between the three types of edit operations (insertion, deletions, and substitutions). Implicitly, our cost function has been uniform, implying that all operations are equally likely. Since substitutions are much more likely, we want to bias our LCS solution with a cost function that prefers substitutions over insertions and deletions.
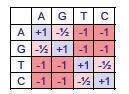
We recast sequence alignment as a special case of the classic Edit-Distance6 problem in computer science (CLRS 366). We add varying penalties for different edit operations to reflect biological occurrences. One biological reasoning for this scoring decision is the probabilities of bases being transcribed incorrectly during polymerization. Of the four nucleotide bases, A and G are purines (larger, two fused rings), while C and T are pyrimidines (smaller, one ring). Thus DNA polymerase7 is much more likely to confuse two purines or two pyrimidines since they are similar in structure. The scoring matrix in Figure 2.6 models the considerations above. Note that the table is symmetric - this supports our time-reversible design.
Calculating the scores implies alternating between the probabilistic interpretation of how often biological events occur and the algorithmic interpretation of assigning a score for every operation. The problem is to the find the least expensive (as per the cost matrix) operation sequence which can transform the initial nucleotide sequence into the final nucleotide sequence.
Complexity of Edit Distance
All algorithms to solve the edit distance between two strings operate in near-polynomial time. In 2015, Backurs and Indyk [? ] published a proof that edit distance cannot be solved faster than \(O\left(n^{2}\right)\) in the general case. This result depends on the Strong Exponential Time Hypothesis (SETH), which states that NP-complete problems cannot be solved in subexponential time in the worse case.
2.3.4 Formulation 4: Varying Gap Cost Models
Biologically, the cost of creating a gap is more expensive than the cost of extending an already created gap. Thus, we could create a model that accounts for this cost variation. There are many such models we could use, including the following:
• Linear gap penalty: Fixed cost for all gaps (same as formulation 3).
• Affine gap penalty: Impose a large initial cost for opening a gap, then a small incremental cost for each gap extension.
• General gap penalty: Allow any cost function. Note this may change the asymptotic runtime of our algorithm.
• Frame-aware gap penalty: Tailor the cost function to take into account disruptions to the coding frame (indels that cause frame-shifts in functional elements generally cause important phenotypic modifications).
2.3.5 Enumeration
Recall that in order to solve the Longest Common Substring formulation, we could simply enumerate all possible alignments, evaluate each one, and select the best. This was because there were only O(n) alignments of the two sequences. Once we allow gaps in our alignment, however, this is no longer the case. It is a known issue that the number of all possible gapped alignments cannot be enumerated (at least when the sequences are lengthy). For example, with two sequences of length 1000, the number of possible alignments exceeds the number of atoms in the universe.
Given a metric to score a given alignment, the simple brute-force algorithm enumerates all possible alignments, computes the score of each one, and picks the alignment with the maximum score. This leads to the question, ‘How many possible alignments are there?’ If you consider only NBAs8 n > m, the number of alignments is
\[\left(\begin{array}{c}
n+m \\
m
\end{array}\right)=\frac{(n+m) !}{n ! m !} \approx \frac{(2 n) !}{(n !)^{2}} \approx \frac{\sqrt{4 \pi n} \frac{(2 n)^{2 n}}{e^{2 n}}}{(\sqrt{\left.2 \pi n \frac{(n)^{n}}{e^{n}}\right)^{2}}}=\frac{2^{2 n}}{\sqrt{\pi n}}\]
This number grows extremely fast, and for values of n as small 30 is too big (> 1017) for this enumeration strategy to be feasible. Thus, using a better algorithm than brute-force is a necessity.
6 Edit-distance or Levenshtein distance is a metric for measuring the amount of difference between two sequences (e.g., the Levenshtein distance applied to two strings represents the minimum number of edits necessary for transforming one string into another).
7 DNA polymerase is an enzyme that helps copy a DNA strand during replication.
8 Non-Boring Alignments, or alignments where gaps are always paired with nucleotides.