
19.5: Disruption of the Cell Cycle Checkpoints Can Cause Cancer
- Page ID
- 16538

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)If a checkpoint fails or if a cell suffers physical damage to chromosomes during cell division, or if it suffers a debilitating somatic mutation in a prior S phase, it may selfdestruct in response to a consequent biochemical anomaly. This is another example of apoptosis. On the other hand, when cells die from external injury, they undergo necrosis, an accidental rather than a programmed death. In the cells shown below, apoptosis or necrosis were chemically induced, followed and identified as apoptotic or necrotic using fluorescent markers (propidium iodide, green; acridine orange, orange).
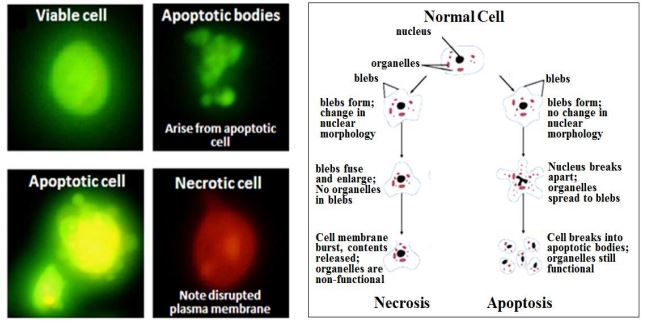
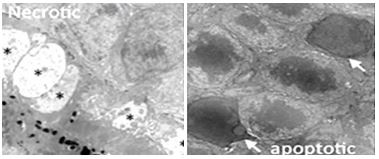
Only green-fluorescing (apoptotic) cells eventually formed apoptotic bodies. In contrast, necrotic (orange-fluorescing) cells lose their plasma membranes, do not form such ‘bodies’ and will eventually disintegrate. (400x magnification). Differences in ultrastructure between necrosis and apoptosis are also seen in electron micrographs of cone and rod cells (left and right, respectively) below. An asterisk indicates the cytoplasmic swelling characteristic of necrotic cone. White arrows point to nuclei characteristic of apoptosis of in rod cells.
As we’ve noted, cycling cells continue to divide until they attain G0 in the terminally differentiated state. Most terminally differentiated cells are cleared by apoptosis when they reach the end of their effective lives, to be replaced by stem cells. We also noted that accidental signaling can bring cells out of G0, leading to renewed cell proliferation. While these cells are obviously abnormal, they are not detected by apoptotic defense mechanisms. Thus, they undergo uncontrolled cell divisions, becoming cancer cells. Likewise, physically damaged or mutated cells may sometimes escape apoptotic clearance. When they do, they may also become cancer cells. Apoptotic clearance and uncontrolled cancer cell proliferation are compared below
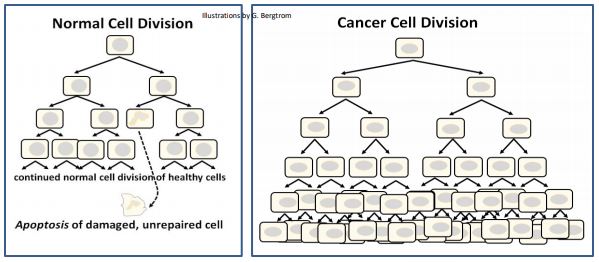
346 Apoptosis (Programmed Cell Death) vs. Necrosis
A. P53 Protein Mediates Normal Cell Cycle Control
Cancerous growth could result if a normal dividing cell should suffer a somatic mutation that disrupts normal cell cycle control. Think an over-expression of cdk for example. Alternatively, imagine cyclin levels in daughter cells that never drop; such cells would never stop cycling.
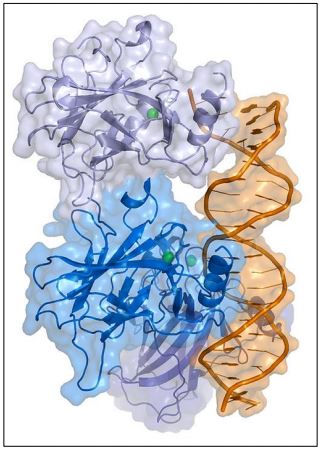
Other possibilities include a cell in G0 that is stimulated to begin cycling again by an inappropriate encounter with a hormone or other signal. If undetected, these anomalies can transform cells to cancer cells. The p53 protein (illustrated below) is a DNA-binding, gene-regulatory protein that detects some of these anomalies and enables dividing cells to repair the damage before proceeding through cell cycle check points…, or failing that, will lead to apoptosis of the cell.
Not surprisingly, mutations in the gene for the P53 protein (called TP53 in humans) are associated with many human cancers (pancreatic, lung, renal cell, breast, etc.). As many as half of human cancers are associated with mutated p53 genes. Thus, p53 is one of a class of tumor suppressor proteins. Studies of humans with a condition known as LFS (Li-Fraumeni syndrome) have at least one mutated p53 allele. The mutation leads to a ~100% lifetime risk of cancer, beginning in childhood. In cultured cells, mutagenized p53 genes exhibit key characteristics of cancer cells, including unregulated cell proliferation and suppression of apoptosis.
1. How p53 Works
The p53 protein is normally bound to an active Mdm2 protein. To enable cell cycle checkpoints, p53-Mdm2 must separate and be kept separate to allow p53 time to act. In dividing cells, physical stress or chemical stress such as DNA damage during cell growth can activate an ATM kinase. ATM kinase in turn, phosphorylates Mdm2, causing it to dissociate from p53. The same kinase also phosphorylates another kinase, Chk2, as well as the now ‘free’ p53. ATM kinase-initiated events are further detailed below.

Each of the proteins and enzymes phosphorylated by the ATM kinase has a role in cell cycle checkpoint function and cell cycle arrest while errors are corrected:
- Now separated from Mdm2, Phospho-p53 actively up-regulates several genes, including the p21 gene.
- The P21 protein binds to cdks; cyclins can’t bind P21-cdks.
- Active Phospho-Chk2 catalyzes cyclin phosphorylation; phospho-cyclins can’t bind to p21-cdks.
- The inability of cyclins to bind cdks specifically blocks the cell cycle between the G1 and S, and the G2-to-M phases.
These kinase-mediated events at cell cycle checkpoints are illustrated below.
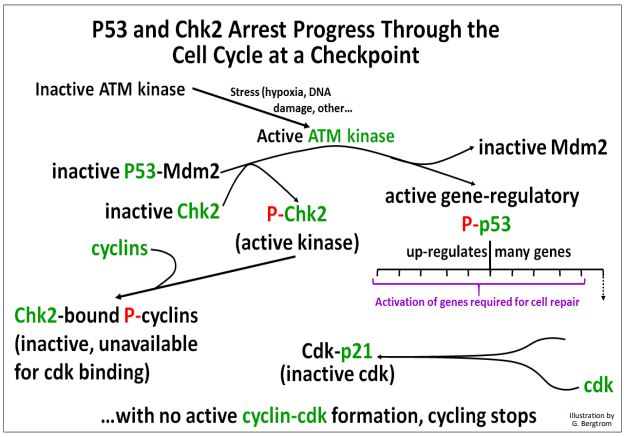
The cell cycle is remains arrested while the cell attempts to finish essential biochemical activities necessary to correct stress-induced or other physical or chemical aberrations before moving on to the next phase of the cycle. If DNA repairs or other corrections are successful, the cell can progress to the next phase.
If not, proteasomes target the Chk2-cyclin complex for degradation. Likewise, any P53 remaining bound to unphosphorylated Mdm2 is also targeted for proteasome destruction. The result is that any cell unable to correct effects of stress or chemical damage, or to repair DNA damage, is target for apoptosis.
The levels and activity of p53 as well as the other proteins discussed above, control both the amount of p53 protein available to respond to cell cycling anomalies, and the responses themselves. Phosphorylation (activation) of p53 not only leads to a rapid arrest of the cell cycle, but also to the activation of genes encoding proteins required for DNA repair and of proteins required for apoptosis (in the event that repair efforts fail). The interactions of p53 with different proteins leading to alternate cell fates are summarized below.

To sum up, p53 suppresses malignant tumor growth either by
- allowing DNA or other cellular repair before resumption of normal cell cycling, preventing unregulated cell divisions; after repair, p53 and other proteins are inactivated and/or destroyed and the cell cycle can resume.
- The inability to repair/correct cell cycling problems sets in motion events leading to apoptosis, thereby also blocking tumorigenesis by killing off damaged cells.
It should be clear now why a mutant p53 that reduces or eliminates p21 protein production or blocks essential DNA repair protein production, will allow damaged cells to enter S and keep them replicating and dividing, transforming such them into cancer cells. In an interesting twist, it seems that compared to humans, few whales or elephants die from cancer, despite having thousands of times more cells than humans. The reason seems to be that, at least for elephants, they have as many as 20 copies (40 alleles) of their p53 genes! Thus a mutation in one allele of one of them may have little effect, while the tumor-repressing effects of the remaining p53 genes prevails. Read about this recent research at Whales and Elephants Don't Get Cancer!
2. The Centrality of p53 Action in Cell Cycle Regulation
Because of its multiple roles in regulating and promoting DNA repair, and in controlling cell cycle checkpoints, p53 has been called “the Guardian of the Genome”! Here is further evidence of this central role.
a) 'Oncogenic Viruses'
Cancer causing viruses include Human Papilloma Virus (HPV), Epstein Barr Virus (EBV), human immunodeficiency virus (HIV), Hepatitis B and C viruses (HBV, HCV), Human herpes virus 8 (HHV-8) and simian virus 40 (SV40).
There is a demonstrated link between SV40, p53 and cancer. SV40 is a viral contaminant of polio vaccines that were used in the 1960s. The virus is tumorigenic in mammals, though an association of SV40 and cancer in humans is unproven. In infected cells, SV40 DNA enters the nucleus where it can integrate into the host cell genome. SV40 infections are usually latent, (i.e., they cause no harm). However, activation can lead to cellular transformation and the growth of malignant sarcomas in muscles as well as tumors in other organs. The RNA polymerase II in infected cells transcribes the SV40 genes, producing proteins that replicate and encapsulate the viral DNA in a membrane to make new viral particles. However, the relatively small SV40 genome does not encode all of the enzymes and factors need for viral DNA replication. The infected cells themselves provide these factors, producing them only during the S phase. At that time, the SV40 large T antigen (made soon after infection) enters the host cell nucleus where it regulates transcription of genes essential to viral replication and viral particle formation. The large T antigen also binds to p53, interfering with transcription of proteins whose genes are regulated by p53. Unable to exercise checkpoint functions, the host cell divides uncontrollably, forming cancerous tumors. Deregulation of the cell cycle by large T antigen ensures progress to the S phase and unregulated co-replication of viral and host cell DNA.
b) p53 and Signal Transduction
Stress can activate signal transduction pathways. For example, mutations affecting the MAPK (MAP kinase) signaling pathway can lead to tumorigenesis. This can be explained by the observation that when activated, the MAPK pathway leads to amplified production of a kinase that phosphorylates p53. Active phospho-p53 in turn augments activation of the MAPK signal transduction pathway. You may recall that MAPK signal transduction typically ends with a mitogenic response.
Another example of p53 interaction is with FAK (focal adhesion kinase) proteins. FAK activity is increased by integrin-mediated signal transduction. Recall that membrane integrins bind fibronectin, contributing to formation of the extracellular matrix, or ECM. Elevated FAK activity participates in the regulation of cell-cell and cell-ECM adhesion at focal adhesion points. Another role for FAK is to bind directly to inactive p53 and increase p53-Mdm2 binding. As we have just seen, persistent p53-Mdm2 is targeted for ubiquitination… and ultimate destruction! In fact, abnormally high levels of FAK are associated with many different tumor cell lines (colon, breast, thyroid, ovarian, melanoma, sarcoma…). These result when p53 is unable properly to activate cell cycle checkpoints.
While the interactions implied here are complex and under active study, these p53 activities certainly confirm its central role as both guardian of the genome and as guardian of cell division.
B. Growth and Behavior of Cancer Cells
Different cancer cell types have different growth and other behavioral properties. You may have heard of slow growing and fast growing cancers. Colon cancers are typically slow growing. Periodic colonoscopies that detect and remove colorectal tumors in middle-age or older people can prevent the disease (although the risks of disease and the procedure itself must be balanced). Pancreatic cancers are fast growing and usually go undetected until they reach an advanced stage. The twin goals of medical research are to detect the different cancers early enough for successful intervention, and of course, to find effective treatments.
A single mutated cell in a tissue can become the growth point of a tumor, essentially a mass of cells cloned from the original mutated one. Benign tumors or growths (for example breast and uterine fibroids in women, or common moles in any of us) stop growing and are not life threatening. They are often surgically removed for the comfort of the patient (or because cells in some otherwise benign tumors may have a potential to become cancerous).
Malignant tumors (also called malignant neoplasms) are cancerous and can grow beyond the boundaries of the tumor itself. When tumor cells are shed they may enter the bloodstream and travel to other parts of the body, the phenomenon called metastasis. Cancer cells that metastasize can become the focal point of new tumor formation in many different tissues. Because cancer cells continue to cycle and replicate their DNA, they can undergo yet more somatic mutations. These further changes can facilitate metastasis and cancer cell growth in different locations in the body.
C. Cancer Treatment Strategies
There are many different kinds of cancers originating in different tissues of the body. They all share the property of uncontrolled cell division, albeit for different molecular and not always well-understood reasons. The two major cancer treatment strategies developed in the 20th century all aim at disrupting replication in some way.
- Radiation therapy relies on the fact that most cells in our bodies do not divide, aiming mutagenic radiation at tumors in the hope that replicating DNA will be mutated at so many sites (i.e., genes) that the tumor cells can no longer survive or replicate properly.
- Chemotherapy is used to attack tumors that do not respond well to radiation or that are not easily be reached by radiation technologies, and to fight cancers that do not even form focused tumors (such as lymphomas and leukemias involving lymph and blood cells). These chemotherapies also aim to derange replication or mitotic activities. For example, recall cordycepin (dideoxyadenosine triphosphate, or ddATP). When present during replication, ddATP is incorporated into a growing DNA chain, after which no additional nucleotides can be added to the DNA strand. That makes ddATP a potent chemotherapeutic disruptor of replication. Taxol is another chemo drug that acts in this case, not by inhibiting S phase replication, but by blocking spindle fiber microtubules from depolymerizing, thus blocking mitotic anaphase and telophase in the latter part of the M and C phases of the cycle. Colchicine (a plant alkaloid) attacks cancer (and other dividing) cells by blocking microtubule formation and therefore preventing spindle fiber formation in mitotic prophase.
These therapies are not effective against all cancers, and of course, they don’t target specific kinds of cancer cells. Their success relies simply on the fact that cancer cells proliferate rapidly and constantly while other cell types do not. Many if not all of the side effects of radiation and chemotherapies result from the damage done to normal dividing cells (e.g., hair follicle cells accounting for hair loss among many cancer patients, depletion of blood cells that fail to be replaced by stem cells in bone marrow).
Much research now is focused on mobilizing the body’s own immune system to create more specific, targeted cancer treatments. In a fascinating bit of history, more than 100 years ago, Dr. William B. Coley injected a terminal cancer patient with streptococcal bacteria, who then emerged tumor-free upon his recovery from the infection (for details, check out The Earliest Cancer Immunotherapy Trials). The phenomenon of “Dr. Coley’s Toxins” was initially thought to be an anti-tumor effect of the bacteria. But by 1948 it was widely attributed to the immune response activated by the infection. In the 1990s, scientists revisited the immune response to cancer, and by the turn of the 21st century, studies of cancer immunotherapy picked up steam (and more substantial research funding!).
Recent animal immunotherapy experiments and human clinical trials are promising. A few immunotherapies have already been approved by the U.S. FDA (Food and Drug Administration). Cancer immunotherapy strategies capitalize on the fact that your body sometimes recognizes cancer cell markers (e.g., cell surface molecules) as foreign, thus mounting an immune defense against those cells. But that response is sometimes not powerful enough to clear new, rapidly dividing cancer cells. Cancer apparently results when the immune response is weak. There are different, sometimes overlapping approaches to cancer immunotherapy. All are based on the fact that cancer cells that have mutated in some way and are producing aberrant proteins that the immune system can see as foreign enough to elicit an immune response, however slight. Some immunotherapies seek to boost that immune response. Others seek isolate or generate unique cancer cell antigens that will immunize a patient when injected with these cancer antigens. Some immunotherapies are summarized in the table on the next page. As you can see from the table, immuno-targeting cancer cells has already proven to be highly effective. In some cases the therapy is an example of personalized medicine, in which treatments are uniquely tailored to you as a patient. Issues with immunotherapies are that
- they are time and labor intensive, and costly to produce.
- while they may ‘cure’ you, they likely won’t not work on someone else.
- like radiation and chemotherapy, immunotherapies come with their own unpleasant and sometimes severe side effects.
A more detailed discussion of cancer immunotherapies is on the cancer.gov website at Cancer Treatment Immunotherpay.

NOTE: The term checkpoint inhibitor in the context of immunotherapies is different than the term checkpoints describing portals to progress through the eukaryotic cell cycle.