
15.2: Make and Screen a cDNA Library
- Page ID
- 16507

The first step in making a cDNA library is to isolate cellular mRNA. This mRNA extract should represent all of the transcripts in the cells at the time of isolation, or the cell’s transcriptome. This term is used by analogy to genome. However, a genome is all of the genetic information of an organism. In contrast, a transcriptome (usually eukaryotic) reflects all of the genes expressed in a given cell type at a moment in time. Reversetranscribed cDNAs from an mRNA extract are also referred to as a transcriptome…, and likewise, a cDNA library. A cDNA library is a tube full of bacterial cells that have taken up (i.e., been transformed with) plasmids recombined with cDNAs. cDNA libraries made from mRNAs taken from different cell types or the same cells grown under different conditions are in effect, different transcriptomes. Each reflects mRNAs transcribed in cells at the moment of their extraction. When cells in a cDNA library are spread out on a nutrient agar petri dish, each cell grows into a colony of cells; each cell in the colony is a clone of a starting cell. cDNA libraries can be used isolate and sequence the DNA encoding a polypeptide that you are studying.
Recall that the mature mRNA in eukaryotic cells has been spliced. This means that cDNAs from eukaryotic cells do not include introns. Introns, as well as sequences of enhancers and other regulatory elements in and surrounding a gene must be studied in genomic libraries, to be discussed later. Here we look at how to make a cDNA library.
A. cDNA Construction
mRNA is only a few percent of a eukaryotic cell; most is rRNA. But that small amount of mRNA can be separated from other cellular RNAs by virtue of their 3’ poly(A) tails. Simply pass a total RNA extract over an oligo-d(T) column (illustrated below).
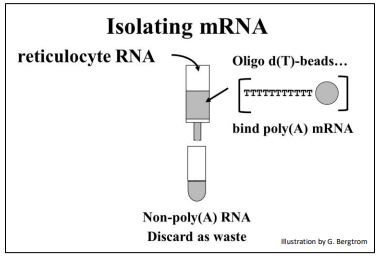
The strings of thymidine (T) can H-bond with the poly(A) tails of mRNAs, tethering them to the column. All RNAs without a 3’ poly(A) tail will flow through the column as waste. A second buffer is passed over the column to destabilize the A-T H-bonds to allow elution of an mRNA fraction. When free’ oligo d(T) is added to the eluted mRNA, it forms H-bonds with the poly(A) tails of the mRNAs, serving as a primer for the synthesis of cDNA copies of the poly(A) mRNAs originally in the cells. Finally, four deoxynucleotide DNA precursors and reverse transcriptase (originally isolated from chicken retrovirus-infected cells) are added to start reverse transcription. The synthesis of a cDNA strand complementary to an mRNA is shown below.
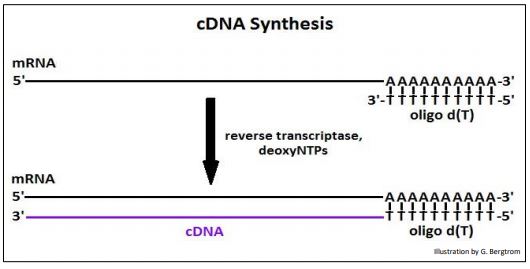
After heating to separate the cDNAs from the mRNAs, the cDNA is replicated to produce double-stranded, or (ds)cDNA, as illustrated below.
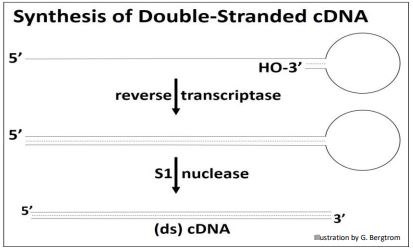
Synthesis of the second cDNA strand is also catalyzed by reverse transcriptase! The enzyme recognizes DNA as well as RNA templates, and has the same 5’-to-3’ DNA polymerizing activity as DNA polymerases. After 2nd cDNA strand synthesis, S1 nuclease (a single-stranded endonuclease originally isolated from an East Asian fungus!) is added to open the loop of the (ds) cDNA structure and trim the rest of the single-stranded DNA. What remains is the (ds) cDNA.
B. Cloning cDNAs into Plasmid Vectors
To understand cDNA cloning and other aspects of making recombinant DNA, we need to talk a bit more about the recombinant DNA tool kit. In addition to reverse transcriptase and S1 nuclease, other necessary enzymes in the ‘kit’ include restriction endonucleases (restriction enzymes) and DNA ligase. The natural function of restriction enzymes in bacteria is to recognize specific restriction site sequences in phage DNA (most often palindromic DNA sequences), hydrolyze it and thus avoid infection.
Restriction enzymes that make a scissors cut through the two strands of the double helix leaves blunt ends. Restriction enzymes that make a staggered cut on each strand at their restriction site leave behind complementary (‘sticky’) ends (below).

If you mix two of double-stranded DNA fragments with the same sticky ends from different sources (e.g., different species), they will form H-bonds at their complementary ends, making it easy to recombine plasmid DNA with (ds)cDNA, that have the same complementary ‘sticky ends’. Using the language of recombinant DNA technologies, let’s look at how plasmid vectors and cDNAs can be made to recombine.
1. Preparing Recombinant Plasmid Vectors Containing cDNA Inserts
Vectors are carrier DNAs engineered to recombine with foreign DNAs of interest. When a recombinant vector with its foreign DNA insert gets into a host cell, it can replicate many copies of itself, enough in fact for easy isolation and study. cDNAs are typically inserted into plasmid vectors that are usually purchased “off-the-shelf”. They can be cut with a restriction enzyme at a suitable location, leaving those sticky ends. On the other hand, it would not do to digest (ds)cDNA with restriction endonucleases since the goal is not to clone cDNA fragments, but entire cDNA molecules. Therefore, it will be necessary to attach linkers to either end of the (ds)cDNAs. Plasmid DNAs and cDNA-linker constructs can then be digested with the same restriction enzyme to produce compatible ‘sticky ends’. Steps in the preparation of vector and (ds)cDNA for recombination are shown below.
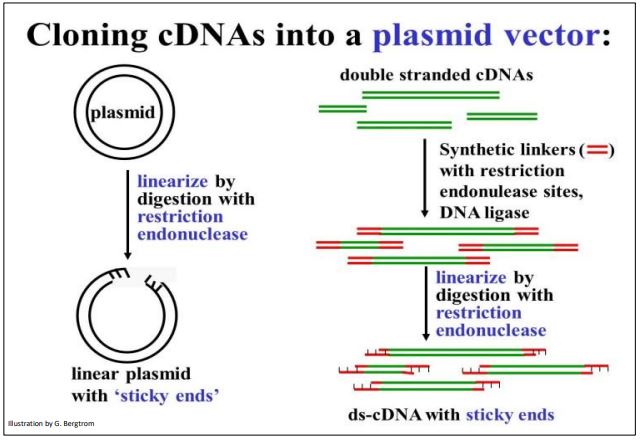
To prepare for recombination, a plasmid vector is digested with a restriction enzyme to open the DNA circle. To have compatible sticky ends, double-stranded cDNAs to be inserted are mixed with linkers and DNA ligase to put a linker DNA at both ends of the (ds) cDNA. DNA ligase is another tool in the recombinant DNA toolkit. Linkers are short, synthetic double-stranded DNA oligomers containing restriction sites recognized and cut by the same restriction enzyme as the plasmid. Once the linkers are attached to the ends of the plasmid DNAs, they are digested with the appropriate restriction enzyme. This leaves both the (ds)cDNAs and the plasmid vectors with complementary sticky ends.
2. Recombining Plasmids and cDNA Inserts and Transforming Host Cells
The next step is to mix the cut plasmids with the digested linker-cDNAs in just the right proportions so that the most of the cDNA (linker) ends will anneal (form Hbonds) with the most of the sticky plasmid ends. Adding DNA ligase to the plasmid/linker-cDNA mixture forms phosphodiester bonds between plasmid and cDNA insert, completing the recombinant circle of DNA, as shown below.
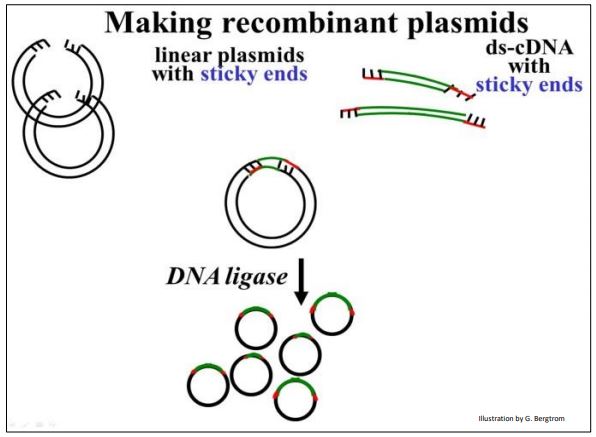
In early cloning experiments, an important consideration was to generate plasmids with only one copy of a given cDNA insert, rather than lots of re-ligated plasmids with no inserts or lots of plasmids with multiple inserts. Using betterengineered vector and linker combinations, this issue became less important.
3. Transforming Host Cells with Recombinant Plasmids
The recombinant DNA molecules are now ready for ‘cloning’. They are added to E. coli (sometimes other host cells) made permeable so that they can be easily transformed. Recall that transformation as defined by Griffith is bacterial uptake of foreign DNA leading to a genetic change. The transforming principle in cloning is the recombinant plasmid! The transformation step is shown below.
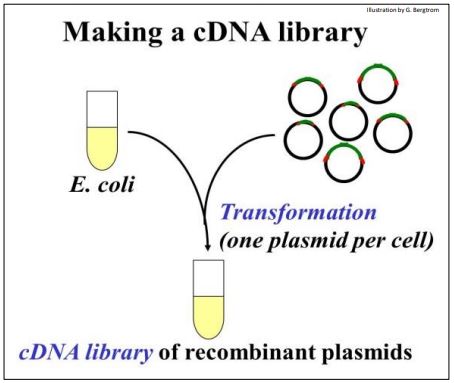
The tube full of transformed cells is the cDNA Library.
After all these treatments, not all plasmid molecules in the mix are recombinant; some cells in the mix haven’t even taken up a plasmid. So when the recombinant cells are plated on agar, how do you tell which of the colonies that grow came from cells that took up a recombinant plasmid? Both the host strain of E. coli and plasmid vectors used these days were further engineered to solve this problem. One such plasmid vector carries an antibiotic resistance gene. In this case, ampicillin-sensitive cells would be transformed with recombinant plasmids containing the resistance gene. When these cells are plated on media containing ampicillin (a form of penicillin), they grow, as illustrated below.

Untransformed cells (cells that failed to take up a plasmid) lack the ampicillin resistance gene and thus, do not grow on ampicillin-medium. But, there is still a question. How can you tell whether the cells that grew were transformed by a recombinant plasmid containing a cDNA insert? It is possible that some of the transformants contain only non-recombinant plasmids that still have the ampicillin resistance gene!
To address this question, plasmids were further engineered with a streptomycin resistance gene. But this antibiotic resistance gene was also engineered to contain restriction enzyme sites in the middle of the gene. Thus, inserting a cDNA in this plasmid would disrupt and inactivate the gene. Here is how this second bit of genetic engineering enabled growth only of cells transformed with a recombinant plasmid containing a cDNA insert. We can tell transformants containing recombinant plasmids apart from those containing non-recombinant plasmids by the technique of replica plating shown (illustrated below).
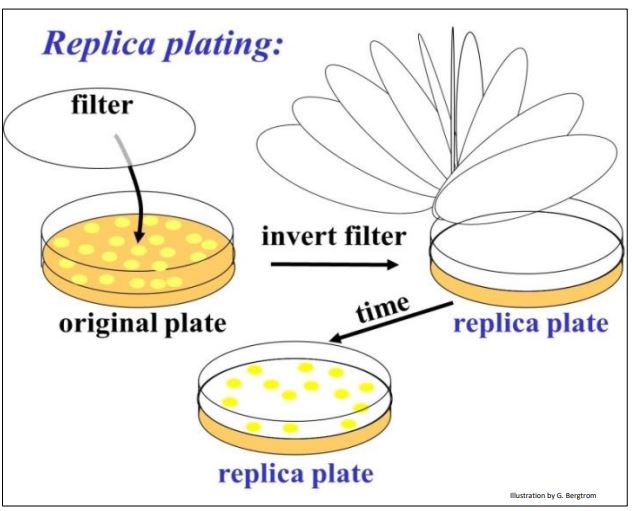
After colonies grow on the ampicillin agar plate, lay a filter over the plate. The filter will pick up a few cells from each colony, in effect becoming a replica (mirror image) of the colonies on the plate. Place the replica filter on a new agar plate containing streptomycin; the new colonies that grow on the filter must be streptomycin-resistant, containing only non-recombinant plasmids. Colonies containing recombinant plasmids, those that did not grow in streptomycin are easily identified on the original ampicillin agar plate. In practice, highly efficient recombination and transformation procedures typically reveal very streptomycinresistant cells (i.e., colonies) after replica plating. In this case, ampicillin-resistant cells constitute a good cDNA library, ready for screening.
4. Identifying Colonies Containing Plasmids with Inserts of Interest
The next step is to screen the colonies from the cDNA library for those containing the specific cDNA that you’re after. This typically begins preparing multiple replica filters like the one above. Remember, these filters are replicas of bacterial cells containing recombinant plasmids that grow on ampicillin but not streptomycin. The number of replica filters that must be screened can be calculated from assumptions and formulas for estimating how many colonies must be screened to represent an entire transcriptome (i.e., the number of different mRNAs in the original cellular mRNA source). Once the requisite number of replica filters are made, they are subjected to in situ lysis to disrupt cell walls and membranes. The result is that the cell contents are released and the DNA is denatured (i.e., becomes single-stranded). The DNA then adheres to the filter in place (in situ, where the colonies were). The result of in situ lysis is a filter with faint traces of the original colony (below).
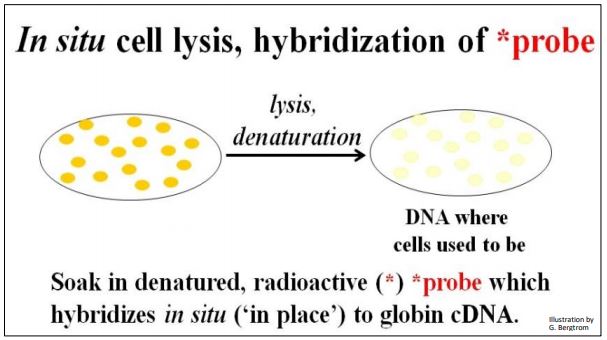
Next, a molecular probe is used to identify DNA containing the sequence of interest. The probe is often a synthetic oligonucleotide whose sequence was inferred from known amino acid sequences. These oligonucleotides are made radioactive and placed in a bag with the filter(s). DNA from cells that contained recombinant plasmids with a cDNA of interest will bind the complementary probe. The results of in situ lysis and hybridization of a radioactive probe to a replica filter are shown below.
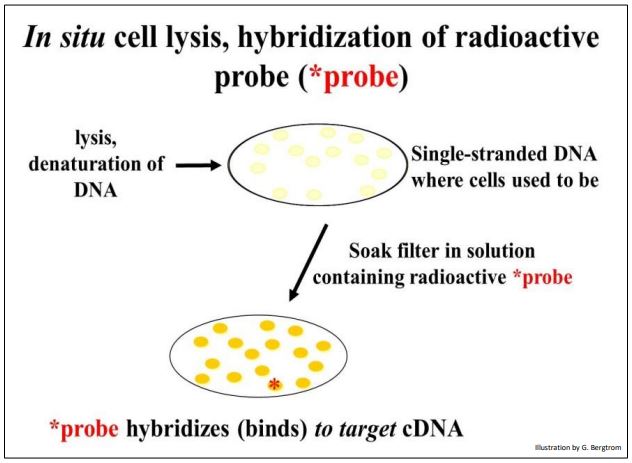
264 Probing a Replica Plate Filter
The filters are rinsed to remove un-bound radioactive oligomer probe, and then placed on X-ray film. After a period of exposure, the film is developed. Black spots will form on the film from radioactive exposure, creating an autoradiograph of the filter. The black spots in the autoradiograph correspond to colonies on a filter that contain a recombinant plasmid with your target cDNA sequence (below).
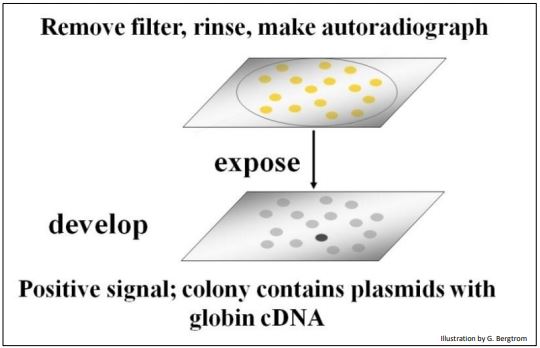
Once a positive clone is identified on the film, the corresponding recombinant colony is located on the original plate. This colony is grown up in a liquid culture and the plasmid DNA is isolated. At that point, the cloned plasmid DNA can be sequenced and the amino acid sequence encoded by its cDNA can be inferred from the genetic code dictionary to verify that the cDNA insert in fact encodes the protein of interest. Once verified as the sequence of interest, a cloned plasmid cDNA can be made radioactive or fluorescent, and used to
- probe for the genes from which they originated.
- identify and quantitate the mRNA even locate the transcripts in the cells.
- quantitatively measure amounts of specific mRNAs.
Isolated plasmid cDNAs can even be expressed in suitable cells to make the encoded protein. These days, diabetics no longer receive pig insulin, but get synthetic human insulin human made from expressed human cDNAs. Moreover, while the introduction of the polymerase chain reaction (PCR, see below) has superseded some uses of cDNAs, they still play a role in genome-level and transcriptome-level studies.