
10.3: Enforcement Activities
- Page ID
- 39535

Civil Enforcement
Regulations must be enforceable to be effective, and the FDA has plenty of tools to encourage compliance. The key to the FDA is product public health and safety. Enforcement proceedings usually take place after an inspection has been deemed OAI. The inspections, through appropriate centers, can be either every two years or as indicated by an issue the FDA has been made aware of. There are two broad categories of enforcement activities; civil and criminal.
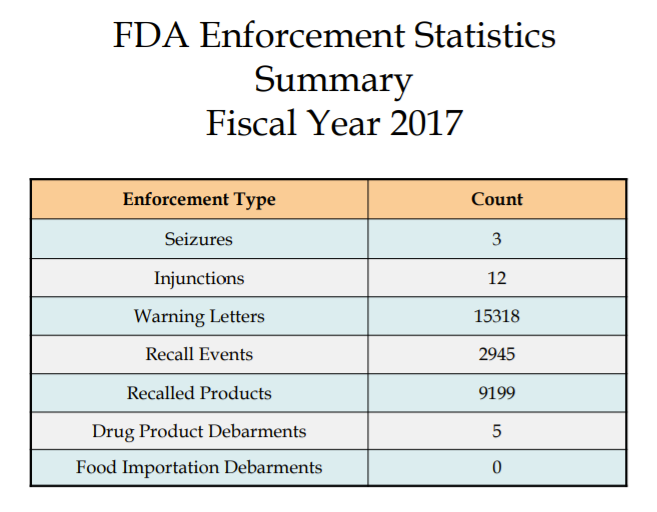
The guidelines to the enforcement actions the FDA takes can be found at the Inspections, Compliance, Enforcement, and Criminal Investigation (ICECI) webpage. The FDA publishes enforcement activities in annual reports to its website. Look around and see what kinds of enforcement activities are most prominent. The 2017 statistics can be found here: https://www.fda.gov/downloads/ICECI/EnforcementActions/UCM592790.pdf
Inspections
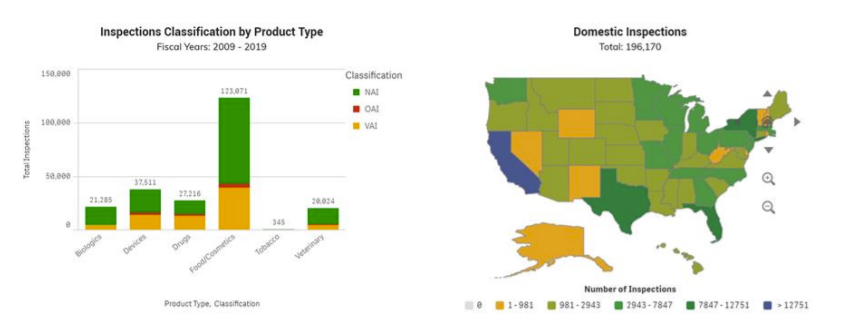
The FDA has the legal authority to inspect pharmaceutical companies, and it can do so unannounced. The guidelines for the inspections are posted to the Inspections, Compliance, Enforcement, and Criminal Investigation (ICECI) webpage. Different inspection guidelines govern different product types. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references
- Inspections Operations Manual: www.fda.gov/inspections-compliance-enforcement-andcriminal-investigations/inspection-references/investigations-operations-manual
- Inspection Guides: www.fda.gov/inspections-compliance-enforcement-and-criminalinvestigations/inspection-references/inspection-guides
- Foreign Inspection Guides: www.fda.gov/inspections-compliance-enforcement-andcriminal-investigations/inspection-references/foreign-inspections
The FDA performs routine (every two years) inspections for compliance with CGMP, GLP, GCP, and Quality System Regulations. They also perform targeted inspections based on other enforcement events such as follow-ups on recall events or warning letters. When the FDA enters a facility for inspection, they provide the inspection request document (FDA 482) as well as provide identification. If they are refused entry, the FDA can obtain a search warrant from a federal court.
The scope of the inspection is primarily based on the reason for the visit and the history of the facility. The only limits the FDA has in inspections relate to financial information, sales data (not related to shipping), personnel data (not related to training), and research data (not related to the approval of products).
During inspections, the FDA looks at seven systems to determine if they are ‘in control’:
- Management responsibility
- Design control
- Corrective and preventative action (CAPA)
- Production and process controls
- Records and document change controls
- Material controls
- Facilities and equipment controls
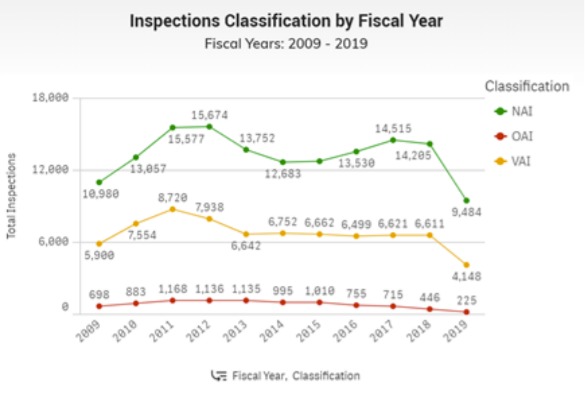
Inspection Classification
After an inspection, the FDA determines if the areas evaluated are in compliance with applicable laws and regulations. FDA and the Inspection Classification Database classifies the inspection by each project area with one of three classifications. The Inspection Classification Database only shows inspections conducted by FDA. www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-classification-database
The FDA conducts careful inspections of facilities that perform nonclinical laboratory studies to determine compliance with Part 58 (Good Laboratory Practice for Nonclinical Laboratory Studies) of Title 21 of the CFR. Nonclinical laboratory studies are experiments in which test articles are studied prospectively in test systems, such as animals, plants, microorganisms under laboratory conditions to determine their safety. Learn more here: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/nonclinical-laboratories-inspected-under-good-laboratory-practices
The three Inspection Classifications displayed are:
- No Action Indicated (NAI) which means no objectionable conditions or practices were found during the inspection (or the objectionable conditions found do not justify further regulatory action),
- Voluntary Action Indicated (VAI) which means objectionable conditions or practices were found, but the agency is not prepared to take or recommend any administrative or regulatory action, or
- Official Action Indicated (OAI), which means regulatory or administrative actions will be recommended.
Inspection Citations: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-citation
Inspections & Form 483
Adulteration and misbranding are the two most common violations found during an inspection. Pharmaceutical facilities and biotechnology companies are periodically inspected by the FDA. Any CGMP violations the inspectors find are noted on forms, called “483’s,” and if no response to the regulatory compliance issues, an official Warning Letter is sent to the company.
A 483 is a result of an inspection and notice of regulatory non-compliance. It is a list of specific items seen at the end of the inspection and is issued by the investigator. After an inspection, the inspector meets with the facilities manager to address as many of the problems before the 483 is written. The inspector offers an opportunity to remedy a situation immediately after the inspection and document compliance and remedies, or future solutions and agreements. After the letter is written and received by the company, the company then has 15 working days to voluntarily respond to the 483 in writing with an action plan. If the 483 issued is not remedied quickly, the FDA can then respond with a warning letter (see below).
It should be noted that the issuance of form 483 does not automatically mean that a firm is not in GMP compliance. Additionally, the length of the form is not a reliable indicator of the seriousness of any observed violations. The form should be viewed critically, and companies that feel they have received a 483 containing questionable observations of GMP deviations should discuss the issue with the inspector, district director, regional director, or even the center of issuance if necessary.
Companies are not required to respond to a 483. However, their response is recommended. If a company receives too many 483s and is not providing the FDA with adequate responses to these forms, they are subjected to a warning letter or more punitive measures as outlined below.
Establishment of Inspection Reports
After the FDA closes an inspection, they release an Establishment of Inspection Report (EIR). It is a formal written report summarizing the findings with supporting evidence. In this report, the FDA classifies the inspection as No Action Indicated (NAI), Voluntary Action Indication (VAI), or Official Action Indicated (OAI). If an OAI is noted, enforcement activities may be listed or be scheduled to come. Inspection Reports and Data: https://datadashboard.fda.gov/ora/cd/inspections.htm
Inspections & Warning Letters
When FDA finds that a manufacturer has significantly violated FDA regulations, the FDA notifies the manufacturer, most often, in the form of a Warning Letter. The Warning Letter identifies the violation and makes clear that the company must correct the problem and provides directions and a timeframe for the company to inform the FDA of its plans for correction. FDA then checks to ensure that the company’s corrections are adequate.
Matters described in FDA warning letters may have been subject to subsequent interaction between the FDA and the recipient of the letter that may have changed the regulatory status of the issues discussed in the letter. A warning letter issued by the FDA is a voluntary informal advisory communication and offers a company the opportunity to remedy the regulatory situation. The overall goal of a warning letter is to encourage voluntary compliance. If the issue does not put the public in immediate danger, a warning letter may be written before other punitive measures. An important aspect of a warning letter is to establish a record of ‘prior notice’ during legal proceedings. However, when issued, a warning letter is published immediately on the FDA website and is shared across regulatory agencies.
- View general FDA warning letters: www.fda.gov/inspections-compliance-enforcementand-criminal-investigations/compliance-actions-and-activities/warning-letters
- View tobacco FDA warning letters: www.fda.gov/inspections-compliance-enforcementand-criminal-investigations/warning-letters/tobacco-retailer-warning-letters
- View drug marketing and advertising warning letters: www.fda.gov/drugs/enforcementactivities-fda/warning-letters-and-notice-violation-letters-pharmaceutical-companies
Warning Letter Closeouts
FDA may issue a Warning Letter close-out letter once they have completed an evaluation and determined that corrective actions were undertaken by a firm in response to a Warning Letter. The corrective actions must have been made and verified by the FDA, usually through a follow-up inspection. If the Warning Letter contains violations that by their nature are not correctable, then no close-out letter will be issued. Future FDA inspections and regulatory activities may further assess the adequacy and sustainability of these corrections. Should violations be observed during a subsequent inspection or through other means, enforcement action may be taken without further notice.
Inspections & Untitled Letters
Untitled letters are the least harsh form of communication with the FDA. Like Warning Letters, the untitled letters can notify a company of a violation that may not reach the threshold of a regulatory issue. Unlike a warning letter, an untitled letter does not include a statement that warns the individual or firm that failure to promptly correct the violation may result in enforcement action. F However, companies are encouraged to address any issues as this does constitute ‘prior notification' and is why you should respond. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/issuance-untitled-letters. For more information on warning and untitled letters: https://www.fda.gov/media/71878/download
Inspections & Press Releases
The FDA can also issue unfavorable press releases or federal register notices. In the event of an egregious defect, the FDA can criminally prosecute, seize materials, and perform injunctions against a company and individuals. (fda.gov). Current press releases: www.fda.gov/inspections-compliance-enforcementand-criminal-investigations/criminal-investigations/press-releases
Seizures
If a company refuses to recall, the FDA can bring a seizure or injunction case against it to address violations even if the products are not defective. Then, they can petition the court for an order that allows federal officials to take possession of “adulterated” drugs and destroy them. This process enables the FDA to immediately prevent a company from distributing defective and potentially harmful drugs to consumers. Both seizure and injunction cases frequently result in court orders that require companies to take many steps to correct violations. These steps may include hiring outside experts to help resolve the problem, writing new procedures, and training employees. In some cases, violations may become criminal cases, allowing the FDA to seek fines and jail time.
Injunctions
If a company violates the FD&C Act, the FDA may file an injunction against the company. This injunction is a civil judicial proceeding and is typically used when a significant health hazard is identified with a product. This FDA may seek a temporary restraining order, a temporary injunction, or permanent injunction. The key here is for the FDA to be able to act quickly to get a product to stop from reaching the customers short of a recall (which the FDA does not have authority in some cases). The FDA also uses this enforcement method if the company has ignored repeated warning letters. If the company addresses the issues, the temporary restraining order can be lifted.
Civil Money Penalties
The FD&C Act, as well as the PHSA, have civil money penalty provisions. Guidance on CMP is provided in 21 CFR 17.2. Look up the CFR on the FDA website, and notice the CMPs are quite harsh! Some penalties exceed a million dollars for aggregate offenses.
Disqualification of Clinical Investigators
FDA regulates scientific studies that are designed to develop evidence to support the safety and effectiveness of investigational drugs (human and animal), biological products, and medical devices. Physicians and other qualified experts ("clinical investigators") who conduct these studies are required to comply with applicable statutes and regulations intended to ensure the integrity of clinical data on which product approvals are based and, for research involving human subjects, to help protect the rights, safety, and welfare of those subjects. In certain situations, in which the FDA alleges a clinical investigator has violated applicable regulations, the FDA can disqualify a clinical investigator and prevent them from providing any clinical data for any product submission. Typically, information on clinical investigators is obtained through the BIMO inspection. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/clinical-investigators-disqualification-proceedings
Debarment
The FDA has the debarment authority of individuals or companies from the drug industry. Debarment means they can no longer manufacturing an approved drug. Debarred companies can no longer manufacture, nor submit any further drug applications. In 2018, the FDA debarred a corporate entity for the first time. Read more here: https://www.federalregister.gov/documents/2018/03/01/2018-04195/meunerie-sawyerville-inc-denial-of-hearing-final-debarment-order.
Debarred people can also be subject to civil money penalties, as well. See a list of debarments on the FDA website. You will notice it is a shockingly long list! https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/fda-debarment-list-drug-product-applications
Criminal Enforcement
Criminal investigations are made by the Office of Criminal Investigations (OCI). It is important to remember when we are discussing FDA regulations on Food & Drugs, we are referring to laws. Breaking laws have many penalties, which can range from fines to jail time. Check out the FDA’s most wanted fugitives here: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/criminal-investigations/office-criminal-investigations-most-wanted-fugitives
Consent Decree
Companies that repeatedly violate CGMP requirements may be forced to make changes via the issuance of a consent decree. The consent decree is signed by the company’s top official, the U.S. Attorney, and the U.S. District Court. The decree is then filed with the court and is later submitted to the FDA. Enforced by the Federal courts, consent decrees typically involve fines, reimbursements to the government for inspection costs, and penalties for noncompliance. Consent decrees can be permanent. However, if a company has complied, it can petition the court to remove the decree.
Test Your Knowledge!
Read here a featured story on the OCI website: "April 4, 2016: Former Carlsbad Resident Jailed for Sale of Unapproved "Energy Wave" Medical Devices."
- What is David Perez accused of doing?
- Why is this against the law? Can you reference the law?
- How long of a sentence did he receive? If he did not receive a plea deal and was found guilty in a court of law, how long would his maximum sentence have been?
- Do you agree with the charge and his sentence? Explain.