
22.1: Overview of Nitrogen Metabolism
- Page ID
- 15178


Introduction
Organic chemistry is usually described as the chemistry of carbon-containing molecules. But isn't that definition a bit carbon centric, especially since the prevalence of oxygen-containing molecules is staggering? What about nitrogen? We live in a dinitrogen-rich atmosphere (80%), and all classes of biomolecules (lipids, carbohydrates, nucleic acids, and proteins) contain nitrogen. Dinitrogen is very stable, given its triple bond and nonpolarity. We rely on a few organisms to fix N2 from the atmosphere to form ammonium (NH4+), which through nitrification and denitrification can form nitrite (NO2-), nitrate (NO2-), nitric oxide (NO), and nitrous oxide (N2O), the latter being a potent greenhouse gas. We'll concentrate on the metabolic fate of amino groups in amino acids and proteins in the next section. Before exploring their fates, look at Figure \(\PageIndex{1}\) which shows an overall view of the biological nitrogen cycle. The study of biochemistry should encompass more than homo sapiens and expand to the ecosystem in which we are such a small but damaging part.

Figure \(\PageIndex{1}\): Nitrogen Cycle
Let's break down the diagram from a biochemical perspective. There are aerobic and anaerobic processes (conducted by bacteria). Nitrogen-containing substances include both inorganic (ammonium, nitrate, nitrite) and organic (amino acids, nucleotides, etc) molecules. The reactions shown are oxidative and reductive (note: the oxidation number of the nitrogen atoms in the molecules is shown in red). Most of the reactions are carried out underground by bacterial and Archaeal microorganisms.
Here are some of the major reactions:
- N2 fixation (a reduction): N2 from the air is converted by bacteria to ammonium (NH4+) by the enzyme nitrogenase of soil prokaryotes. The energetically disfavored reaction requires lots of ATPs. Ammonium once made can then be taken up by primary producers like plants and incorporated into biomolecules such as amino acids, which animals consume. For those who may still believe that people have marginal effects on our biosphere, consider this. We may soon fix more N2 to NH3 through the industrial Born-Haber reaction (used for fertilizer and explosive productions) that is all made by the biosphere. Much of the nitrogen in use comes from the Born-Haber reaction. The excess NH4+ (upwards of 50%) produced industrially and which enters the soil in fertilizers (mostly as NH4NO3) has overwhelmed nature's ability to balance the nitrogen cycle and is not taken up by plants. It is metabolized by microorganisms to nitrite and nitrate.
- Nitrification: Ammonium is converted to nitrite by ammonia-oxidizing aerobic microorganisms and further to nitrate by a separate group of nitrite-oxidizing aerobic bacteria. Here are the reactions (Rx 1 and 2) to produce nitrate through a hydroxylamine intermediate, followed by the formation of nitrate (Rx 3).
NH3 + O2 + 2e- → NH2OH + H2O Rx 1
NH2OH + H2O → NO2- + 5H+ + 4e- Rx 2
NO2- + 1/2 O2 → NO3- Rx 3
These added ions exceed soil capacity and end up runoff water, polluting our rivers and lakes.
- Denitrification: This anaerobic reaction pathway reproduces N2 from nitrate Here is the net reaction:
2NO3- + 10e- + 12H+ → 2N2 + 6H2O
- Anammox reaction: This more recently discovered bacterial anaerobic reaction pathway converts ammonium and nitrate to N2. Here is the net reaction
NO2- + NH3+ → N2 + 2H2O
- Ammonification (not to be confused with mummification) occurs when plants and animals decompose, which returns ammonium to the soil for reuse by plants and microbes.
These reactions are shown in the abbreviated Nitrogen Cycle shown in Figure \(\PageIndex{2}\).

Figure \(\PageIndex{2}\): Abbreviated Nitrogen Cycle
Nitrogen metabolites are nutrients for plants and perhaps the most important nutrients in the regulation of plant growth (primary productivity) and in regulating life diversity in the biosphere. All living organisms require feedstocks to produce energy and as substrates for biosynthetic reactions. Which is used depends on the organism. Plants are primary producers so they use their synthesized carbohydrates for both energy production and biosynthesis. For carnivores, proteins and their derived amino acids are the source of energy (through oxidation) and serve as biosynthetic precursors. For omnivorous organisms, the source of energy depends on the "fed" state. With abundant food resources, carbohydrates, and lipids are the source of energy. Unlike carbohydrates and lipids, which can be stored as glycogen and triacylglycerols for future use, excess protein, and their associated amino acids can not be stored, so amino acids can be eliminated or used for oxidative energy.
In the fed state, carbohydrates are the main source, while in the unfed state, lipids take a predominant role. Under starving conditions, the organism's own proteins are broken down and used for oxidative energy production and for any biosynthesis that remains. In diseased states like diabetes, which can be likened to a starving state in the presence of abundant carbohydrates, both lipids and amino acids become the sources of energy.
How are amino acids in animals oxidatively metabolized? Many pathways could be used to do so but it would seem logical that NH4+ would be removed and the carbons in the remaining molecule would eventually enter glycolysis or the TCA cycle in the form of ketoacids. NH4+ is toxic in high concentrations. Ammonium is not oxidized to nitrite or nitrates in humans as occurs in the soil by microorganisms. It can be recycled back into nucleotides or amino acids, and excess amounts are eliminated from the organism. Both processes must be highly controlled. We will turn out attention to the oxidation of amino acids in the next section.
Nitrogenase: An Introduction
Beauty is in the eye of the beholder.
As the domain of biochemistry covers the entire biological world, the extent of coverage of a given topic in textbooks can depend, in part, on the interest and experiences of the author(s) presenting the material. Is relevance a metric that should determine coverage? If so, books focused on human or medical biochemistry would surely omit photosynthesis. If topics are selected based on their importance for life, then photosynthesis must surely be covered. If so, then nitrogenase must also be included. If the degree of chemical difficulty for a chemical reaction and the amazing eloquence of the evolved biochemistry solution is considered, then both photosynthesis and nitrogen fixation must be presented. Even though nitrogen fixation is a reductive reaction, it shares strong similarities with the oxygen-evolving complex of photosynthesis. They catalyze enormously important redox reactions that involve an abundant atmospheric gas using a very complicated and unique inorganic metallic cofactor that evolution has selected as uniquely suited for the job.
Every first-year student of chemistry can draw the Lewis structure of dinitrogen, N2, which contains a triple bond and a lone pair on each nitrogen. If Lewis structures speak to them, they should be able to state that the triple bond makes N2 extraordinarily stable, thus explaining why we can breathe an atmosphere containing 80% N2 and not die. If they have taken biology, they are also aware that very few biological organisms can utilize N2 as a substrate, as this requires breaking bonds between the nitrogen atoms, a chemical process reserved for nitrogen “fixing” bacteria found in rhizomes of certain plants. Lastly, they probably memorized that high pressure and temperature are needed in the Haber-Bosch process used to convert N2 and H2 to ammonia, NH3. As with any scientific advance, the Haber-Bosch process has brought both harm (it's used for explosive weapons) and good (fertilizers). This process now fixes enough N2 in the form of fertilizers to support half of the world’s population, with nitrogenase supporting the rest. Efforts are underway to genetically modify plants to make nitrogenase, eliminating the need for fertilizers but perhaps creating unforeseen problems of its own.
You might be surprised to find out that at room temperature the equilibrium constant favors ammonia formation, hence ΔG0 < 0. The reaction is favored enthalpically as it is exothermic at room temperature. It is disfavored entropically as should be evident from the balanced equation:
\[\ce{N2(g) + 3H2(g) → 2 NH3(g)}. \nonumber \]
The thermodynamic parameters for the reaction (per mol) are ΔH° = –46.2 kJ, ΔS° = –389 J K–1, and ΔG° = –16.4 kJ at 298 K
The entropy is negative since the reaction proceeds from 4 molecules to two molecules. From an enthalpy perspective, if you raise the temperature of an exothermic reaction, you drive it in a reverse direction. If you increase the pressure, you shift the equilibrium to the side that has the fewest number of molecules.
If the reaction is favored thermodynamically at room temperature, why doesn’t it proceed? This story sounds familiar as this same descriptor applies to the oxidation of organic molecules with dioxygen. There we showed using MO theory that the reaction is kinetically slow. Same with NH3 formation. A superficial way to see this is that we must break bonds in the stable N2 to start the reaction, leading to a high activation energy, and making the reaction kinetics sluggish.
One could jump-start the reaction by raising the temperature, but that would slow an exothermic reaction. The Keq (or KD) and ΔG0 are functions of temperature and for this reaction, the reaction becomes disfavored at higher temperatures. The solution Haber found was high pressure, forcing the reaction to the side that has fewer molecules of gas, and high temperature to overcome the activation energy barrier and make the reaction kinetically feasible. A complex metal catalyst (magnetite - Fe3O4 -with metal oxides like CaO and Al2O3 which prevent reduction of the Fe with H2) provides an absorptive surface to bring reagents together and facilitate bond breaking in H2 and N2.
In photosynthesis, the oxygen-evolving complex (OEC) with Mn, Fe, S, and Ca is used to oxidize another very stable and ubiquitous molecule, H2O. Now we explore the amazing mechanisms behind the nitrogenase complex which fixes N2 to form NH3 in a reductive fashion.
What might be needed to drive this reaction biologically? You might surmise the list to include:
- a source of energy, most likely ATP, to facilitate this complex reaction;
- a source of electrons as the N atoms move from an oxidation state of 0 in elemental N to 3- in NH3; this source turns out to be a protein called flavodoxin or ferredoxin. Of course, these electrons also have interesting sources before they were in the electron carriers of these proteins;
- some pretty amazing metal centers to accept and donate electrons in a controlled way; these centers are mostly FeS clusters with an additional cluster containing molybdenum (Mo). The clusters are named F, P, and M
- a source of hydrogen; you might have guessed correctly that it’s not H2 gas (from where would that come?), but H+ ions which are pretty ubiquitously available.
- a net reaction that is different that the Haber-Bausch process (N2 + 3H2 → 2 NH3).
Here is the actual reaction catalyzed by nitrogenase:
\[\ce{N2 + 8e^{-} + 16ATP + 8H^{+} → 2NH3 + H2 + 16ADP + 16P_i}. \nonumber \]
Let’s think a bit about the reaction. As electrons are added the attraction between the nitrogen atoms must decrease. Eventually, bonds between them must be broken. Protons could be easily added to maintain charge neutrality. A basic mechanism might involve intermediates as shown in Figure \(\PageIndex{3}\).

Figure \(\PageIndex{3}\): Possible intermediates in the conversion of dinitrogen to ammonia by nitrogenase
Nitrogenase can also other small molecules with triple bonds, including C=O: and H-C=C-H.
The Structure of Nitrogenase
Nitrogenase is a multiprotein complex in which the functional biological unit is built from two sets of the following dimeric structures:
- a homodimer of subunits, E and F, which have binding sites for the mobile carrier of electrons (the protein ferredoxin or flavodoxin), ATP, and an FeS cofactor (4Fe-4S, called the F cluster) which accept electrons. These subunits are hence called the nitrogenase reductase subunits
- a heterodimer of alpha and beta subunits. The a (alpha chain) binds the 8Fe-7S F cluster and the Fe-S-Mo M cluster. These subunits comprise the (di)nitrogenase catalytic subunits. The iron-molybdenum M cluster is in the α subunit and is where N2 reduction occurs. The P-cluster is between the α and β subunits and facilitates electron flow between the Fe-protein (F cluster) and FeMo-cofactor (M cluster)
For clarity, one-half of the overall structure of the protein complex with bound ATP and metal centers is shown in Figure \(\PageIndex{4}\).

Figure \(\PageIndex{4}\): Nitrogenase structure (4wzb)
This half-structure consists of a homodimer of the reductase monomers and a heterodimer of nitrogenase subunits.
Figure \(\PageIndex{5}\) shows an interactive iCn3D model of the half structure of nitrogenase complex from Azotobacter vinelandii (4WZB) (long load).
.png?revision=1&size=bestfit&width=426&height=362)
 Figure \(\PageIndex{5}\): Nitrogenase complex from Azotobacter vinelandii (4WZB). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zPNjfPRwq8MSeA (long load))
Figure \(\PageIndex{5}\): Nitrogenase complex from Azotobacter vinelandii (4WZB). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zPNjfPRwq8MSeA (long load))
The structure is color-coded in a fashion similar to Figure 4. The F cluster is labeled as SF4, the P cluster as CLF (FE(8)-S(7) cluster), and the M cluster as ICS (iron-sulfur-molybdenum cluster with interstitial carbon).
The reductase subunits (called Av2 in Azotobacter vinelandii), accept electrons from ferredoxin and is where the ATP analog, ACP (phosphomethylphosphonic acid adenylate ester), and the 4Fe-4S F cluster is bound. The nitrogenase subunits (called Av1 in Azotobacter vinelandii) convert N2 to NH3 and is where the 8Fe-7S P cluster, FeMo (C Fe7 Mo S9) M Cluster is bound.
An enhanced view of the bound cofactors and ATP is shown in the same spatial orientation in Figure \(\PageIndex{6}\)below.

Figure \(\PageIndex{6}\): Bound cofactor metal clusters and ATP in nitrogenase
You can easily image the direction of the flow of electrons from the F cluster to the P cluster to the M cluster.
The metal centers are shown in Figure \(\PageIndex{7}\) in more detail in both line and space fill views.
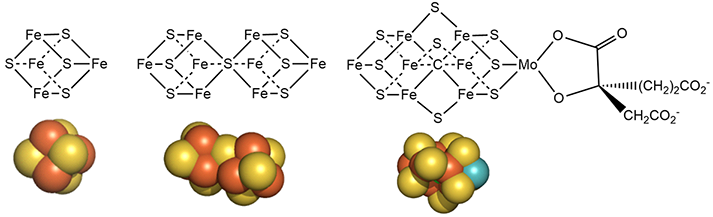
Figure \(\PageIndex{7}\): Detailed structures of the metal cofactors in nitrogenase
Mo is bound to 3 sulfur ions and oxygen from 3-hydroxy-3-carboxy-adipic acid as shown in Figure 7 above.
The M cluster has an interstitial carbide ion that derives from -CH3 attached to the sulfur of S-adenosyl-methionine (SAM) allowing the carbide to be labeled with either 13C or 14C for mechanistic studies. These labeled carbides are not exchanged or used as a substrate when the enzyme undergoes catalytic turnover. Hence it seems that the carbide probably just stabilizes the M cluster. it won't be shown in the figures below showing more detailed mechanisms.
We'll focus on two features of the reaction mechanism, ATP hydrolysis and the flow of electrons and protons to N2 as it is reduced to NH3.
ATP hydrolysis
ATP binds in the reductase subunit (AV2) where ferredoxin brings in electrons, and where the F metal cluster is bound. Much as GTP hydrolysis controls conformational change and subunit dissociation in the heterotrimeric Gαβγ in signal transductions, ATP hydrolysis in reductase (AV2) subunits, drives not only electron transfer but dissociation/reassociation of the reductase and nitrogenase catalytic subunits. It appears that 2 ATPs are hydrolyzed per electron transferred from the F cluster to the Mo M cluster. Since the oxidation numbers of N are 0 and -3 in N2 and NH3, respectively, sequential rounds of ATP hydrolysis and dissociation/reassociation occur. Note that the Fe-protein hydrolyzes ATP only when bound to the MoFe-protein.
The overall reaction involves the reduction of N2 to two molecules of NH3 at the FeMo cofactor. It involves a reductive elimination of hydrides that are bridged by Fe ions (Fe-H-Fe) in a reaction that also makes H2 as a by-product. We'll describe this complex reaction next.
Nitrogenase Reaction: Part 1 - Addition of Electrons and Protons
The sequential path of electrons from the reductase subunit containing the F cluster to the P and M clusters in the nitrogenase subunit should be apparent from the figures above. We will concentrate on the binding of N2 and how it receives electrons from the M cluster. Figure \(\PageIndex{8}\) shows the FeMo-cofactor and some adjacent amino acid residues. Mo is labeled but not shown in spacefill. HCA is a bound molecule of 3-hydroxy-3-carboxy-adipic acid, which interacts with Mo. The carbide is shown in the middle as the green sphere and labeled CX.
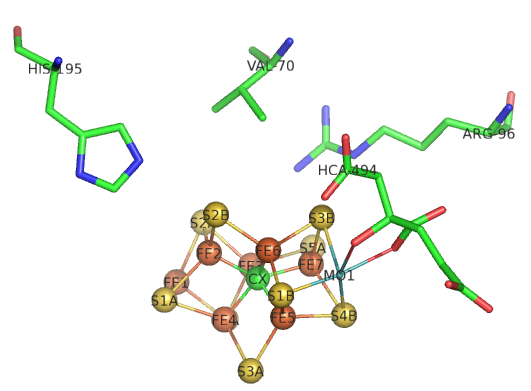
Figure \(\PageIndex{8}\): Side chains near the M cluster
If Val 70 is mutated to Ile, a substrate appears not to access the cluster suggesting that N2 may interact with the top part of the structure with the residues shown acting as gatekeepers. His side chains are often found at enzyme active sites so you might expect His 195 to be a general acid/base. Mutations lead to drastic losses in the reduction of N2. His 195 is involved in hydrogen bonds to sulfur S2B and bridges Fe2 and Fe3 in Figure 8, where reduction of N2 likely occurs. If His 195 moves, it can form short H bonds between the imidazole N and an H bond to HFe2. If the ring is rotated 1800, no proton transfer occurs from the surface. It appears that His195 might be involved in the first N2 protonation event.
The Lowe and Thorneley (LT) model has been proposed as a mechanism for dinitrogen reduction. In this model, an electron and proton are added to the oxidized form of the enzyme (Eo) to produce E1. This is repeated 3 more times to form sequentially, E2, E3, and E4. Only then does N2 bind and the reduction of N2 occurs. Two of the added electrons are accepted by H+ ions which form H2, which is liberated on N2 binding. Hence only 6 electrons are added to the actual N2 molecule, in agreement with the change in oxidation numbers discussed above. The Lowe and Throneley model is shown in Figure \(\PageIndex{9}\).
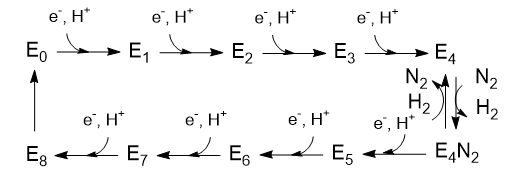
Figure \(\PageIndex{9}\): Lowe and Throneley model for electron and proton additions in nitrogenase
The crystal structure shows 2 ATP analogs bound to the reductase subunit. The stoichiometry of the reaction shows 16 ATP used. Simple math suggests that 2 ATP are cleaved to support the entry of one electron into the complex, assuming 8 transferred electrons (6 to N2 and 2 to 2 protons to form H2).
Part 1 - E1-E4: A potential structure for the E4 intermediate is shown in Figure \(\PageIndex{10}\).
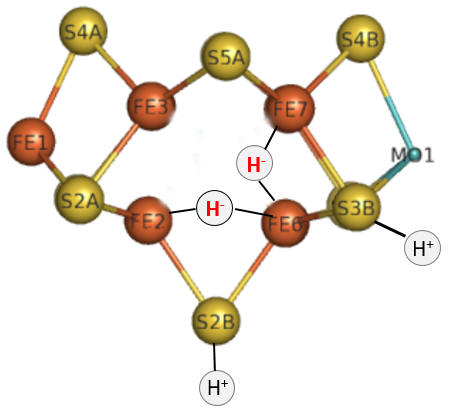
Figure \(\PageIndex{10}\): The E4 Janus intermediate in the reduction of N2.
Note the central carbide is not shown. This is often called the Janus intermediate as it is halfway through the catalytic cycle. It is named for Janus, the Roman god of beginnings and transitions, and has been ascribed to gates, doors, doorways, and passages. Janus is typically shown with two faces, one looking to the future and one to the past (image below: DOI:10.1590/2177-6709.21.1.018-023.oin. License CC BY 4.0 Creative Commons Attribution 4.0 International)

The hydrides bridge 2 Fe ions so these are examples of three-center, two-electron bonds. The H+ ions in Figure 10 balance the charge from the hydrides.
How does this reaction occur? We must look to organometallic chemistry to help us understand the mechanism of this and subsequent steps. Hydride equivalents have been added to the metals, associated with the oxidation of metal ions in the center. This particular reaction is called an oxidative addition. Presumably, the sulfur ions act as Lewis bases as they gain protons from a Lewis acid, probably His 195.
Oxidative addition reactions
Figure \(\PageIndex{11}\) shows oxidative additions to metal centers for three different types of reactants.

Figure \(\PageIndex{11}\): Three different types of oxidative addition reactions (after Schaller, http://employees.csbsju.edu/cschaller/ROBI1.htm)
In oxidative insertion, the oxidation state of the metal ion increases, hence the name oxidative. The hydrogens are now hydrides. Note the example for insertion of H2, an example similar to the proposed hydride additions to the M cluster. Oxidative reduction occurs most readily when the two oxidation states of the metal ion are stable. It is likewise favored for metal centers that are not sterically hindered (makes sense if A-B is to be added) and if A-B has a low bond dissociation energy.
One way to study reaction intermediates is to trap them. If N2 can't access the binding site and the temperature is reduced, the accumulated hydrides (and for charge balance the H+s) in E4 might leave in the opposite reaction, reductive elimination, which we will discuss below, as the reaction goes back to E1. In the elimination, they could form H2 as metal gains back electrons in a reduction. The Val70Ile discussed above would allow an intermediate to be trapped.
Nitrogenase Reaction: Part 2 - Reduction of N2
Step E4 to E5 seems a bit bizarre as H2 gas is released. This would seem to waste ATP but we should trust evolution has led to this mechanism for a reason. This mechanism, the reverse of oxidative addition, is another classic organometallic reaction, reductive elimination.
Reductive elimination reactions
In this reaction, a molecule is eliminated or expelled from the complex as the metal ion is reduced and adds two electrons. Figure \(\PageIndex{12}\) shows reductive elimination. Reductive elimination occurs most readily in higher oxidation state metal centers which can be stabilized on reduction. It occurs most readily from electron-rich ligands and if the other surrounding ligands are bulky. The dissociating species must also be cis to each other in the transition metal complex so they can form a bond with each other when they leave.
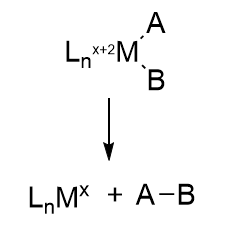
Figure \(\PageIndex{12}\): Reductive Elimination reaction (after Schaller, ibid)
Oxidative addition and reductive elimination (OA/RE) reactions at metal centers are often coupled together in organometallic catalytic cycles, in the same way as a histidine can act as a general acid and then accept a proton back as a general acid to complete the catalytic cycle. In the OA/RE reactions at metal centers, some rearrangements or other modifications can also occur. Think about it. The FeS clusters must return to their original oxidation state after the complete LT cycle. We will encounter another organometallic reaction after the addition of N2, migratory insertion, in the second half of the reaction. Another advantage of coupling OA/RE is that the positive charge or oxidation state on the transition metal complex does not get too high, which is unstable. Making a cation with a positive charge more positive becomes more difficult, much as removing a second proton from a polyprotic acid is more difficult than removing the first (as reflected in the higher pKa for removal of the second proton).
Is H2(g) really released? To study this, investigators have used alternative substrates like acetylene, HC=CH (similar to N=N), in the presence of D2 and N2 in an aqueous system. It helps to see Figure \(\PageIndex{9}\) again.
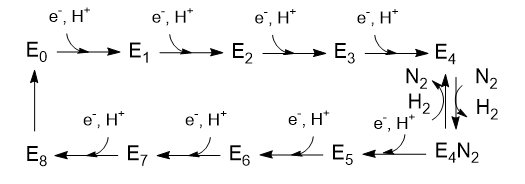
Figure \(\PageIndex{9}\): Lowe and Throneley model for electron and proton additions in nitrogenase
The acetylene was reduced and formed C2H2D2 and C2H3D. Hence E4 must have had 2 Ds in it, and E2 probably 1. These results support the reversible reductive elimination mechanism for the E4 to E4:N2 reaction above. Previously it had been shown that H+ are reduced by D2 in the presence of D2 and N2 in an aqueous system, so these results are consistent. In additional support, deuterium from D2 is not incorporated into products (C2H2D2, C2H3D, or HD) in the absence of N2.
Let's return for a moment to the bridging hydrides as shown again Figure \(\PageIndex{10}\).
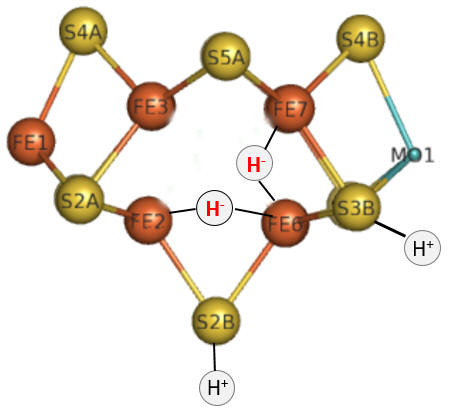
Figure \(\PageIndex{10}\): The E4 Janus intermediate in the reduction of N2
To summarize, it appears likely that the reductive elimination of the two proximal bridging hydrides is the mechanism for the formation of H2. The bridging hydrides, which are strong bases, are much less likely to be protonated than if the hydrides were terminal. A simple and competing protonation reaction could form H2 as well, and if that occurred the reducing equivalents of the bridging hydrides would be lost. Hence the bridging hydrides (share by two metal centers) are more stable and hence can "wait" for the incoming N2 reactants before their reducing equivalents are lost. They may convert to terminal hydrides eventually to facilitate substrate reduction (hydrogenation).
The Janus intermediate E4 is now in a position to bind N2 and release H2. For each H- that binds to the M cluster, two H+s bind to the M cluster sulfides for electrostatic stabilization.
Migratory Insertions
We need to consider one last common type of reaction at a metal center, migratory insertions (MI). In a MI reaction, a group attached to a metal ion center is transferred to another group attached to the same metal. Figure \(\PageIndex{13}\) shows four examples of MI reactions.

Figure \(\PageIndex{13}\): Migratory insertion reactions at metal centers
Panel (a) shows a generic MI reaction. Panel (b) shows the interaction of carbon monoxide, :C=O: (isoelectronic and analogous to :N=N:), with a metal center. Panel (c) shows how a hydride could engage in 1:1 insertion as it shifts and covalently bonds to the first atom of another ligand bound to the metal center. Note that metal does not have to have a negative charge. This could theoretically be important for the reduction of N2 bound as a ligand through a coordinate covalent (dative) bond to the metal. Panel (d) shows the migration of an alkyl group.
We will see that the MI reaction is involved in adding Hs to N2 starting not with N2 but with the N2H2 stage as the intermediates insert into an Fe-H bond. N2 is not reactive to the insertion of a hydride as is carbon monoxide, CO, which provides a positive oxygen to facilitate electron flow during the insertion. In addition, the oxygen become neutral after the reaction.
This offers a great explanation for how Nature chose the FeMo cluster for nitrogen fixation. The interaction of two hydrides requires a 4 Fe face (coordinated with the carbon) that allows for the storage of reducing equivalents for the initial reduction of N2. The large M cluster is more stable and effectively held together by a central carbide anion. This also allows the metal centers to never change their oxidation state by more than 1 charge unit.
The source of the protons to form N2H2 comes directly from the two H2 attached to the two sulfurs as they can't come from the hydrides which are released as H2. The seeming wasteful reductive elimination of H2 and energy is required to allow the kinetically unreactive N2 molecule to bind to the reduced and activate 4Fe face which is also electrostatically facilitated by the 2 bound protons in what has been called a push (reductive)-pull (protonation) reaction. This first step in N2 reduction to N2H2 is the hardest.
Oxidation States of Nitrogenase Fe centers
First Half: It would be difficult to assign specific oxidation states to each Fe ion in the M complex. Instead, we can assign relative changes in the oxidation states as the reaction proceeds from E0 to E4. In each step of the LT model, 1 electron is added. We will first assign this to an average Fe ion, M0, with an arbitrarily assigned oxidation state of 0. On the addition of 1 electron, the oxidation state would go from M0 to M-1 as the metal is reduced. The M-1 state is then oxidized as an electron is transferred to H+, and when 2 electrons are transferred, a single H- is made. The diagram in Figure \(\PageIndex{14}\) shows the change in oxidation state in going from E0 to E4.

Figure \(\PageIndex{14}\): Change in oxidation state in going from E0 to E4
The red boxes highlight thermodynamic cycle-like steps which show how changes in the redox state of the Fe ions (M) could be visualized. Note that in going from E0 to E4, the actual oxidation state of M changes from 0 to +1 to 0 to +1 and back to 0. That is quite amazing given that 4 electrons have been added. Note also that in the red box going from step E0 to E1, M goes from -1 to +1 which corresponds to our description of an oxidative addition when the metal center loses two electrons. This mechanism shows that nitrogenase could be considered a "hydride storage device".
Second Half (facing forward to production NH3):
How does N2 initially interact with E4? It must depend on how the hydrides are released as H2, which evidence shows occurs by reductive elimination (re) and not hydride protonation (hp). On addition, the N2 very quickly is converted to diazene, HN=NH, with the departing H2 taking with it 2 H+s and 2 electrons (or reducing equivalents). These events could occur as shown in Figure \(\PageIndex{15}\).

Figure \(\PageIndex{15}\): Reaction mechanism for the formation of N2H2
Now, with N2 bound as diazene (N2H2) and H2 released, the rest of the reaction could occur as shown below. One new step, a migratory insertion, is shown in Figure \(\PageIndex{16}\).

Figure \(\PageIndex{16}\): Reaction mechanism for the conversion N2H2 to NH3.
The two halves of the reaction are similar with bridging hydrides utilized - the E4 Janus intermediate links the two halves together.