
12.3: The Chemistry and Biochemistry of Dioxygen
- Page ID
- 21163


The History of Oxygen
Oxygen may be considered one of the most important elements in chemistry. Not counting hydrocarbons, there is a greater diversity of molecules with oxygen than with carbon. Given its role in the molecular world, very little time is spent on the chemistry of oxygen in undergraduate chemistry classes. Why is oxygen so special?
Oxygen reacts with atoms of all elements except the Noble gases to form molecules. One of the most important molecules of course, from a biological sense, is water. It :
- provides a perfect solvent for biomolecules
- moderates the earth's climate
- is the source of almost all the dioxygen in the air
From a chemical point of view, water is a(n):
- nucleophile and electrophile
- acid and base
- oxidizing agent and reducing agent
- a protic solvent that can form H-bonds
The formation of the earth and the development of life:
The gaseous and dusty environment from which the earth was formed contained metals and water, which as you remember from introductory chemistry, can react to form hydrogen gas. H2 reacts with nonmetals (under various conditions of temperature and pressure) to form H2S, HCl, CH4, and NH3 which contributed to the reducing nature of the early atmosphere. This kept the transition metals in their lowest oxidation states. Many metals, including the coinage metals (Cu, Ag, and Au) and the platinum group (Ru, Rh, Pd, Pt) were stable in elemental form.
Then, around 2.7-2.8 billion years ago, photosynthetic organisms (blue/green algae- also called cyanobacteria) developed which could oxidize water to form dioxygen. Oxygen was generally unavailable for redox chemistry before then as photosynthesis, the process that would evolve to oxidize water to produce dioxygen, was unavailable. Remember that to oxidize water to dioxygen, itself a strong oxidizing agent, requires a stronger oxidizing agent than dioxygen and lots of energy. Fossilized remains of cyanobacteria are found in stromatolites. Using knowledge of how atmospheric oxygen can alter the chemistry of different sulfur isotopes of SO2, it has been shown that O2 did not exist in the atmosphere as a whole above 1 ppm earlier than 2.4 billion years ago, although there might have been isolated pockets with higher concentrations. After that, it rose, presumably as a result of cyanobacteria. Before this time, bacteria oxidized a similar molecule, H2S to form elemental sulfur. It could do this through the photosynthetic reduction of CO2 by H2S. Volcanic gases like H2 might have kept oxygen levels from rising between 2.7 billion years ago and 2.4 billion years ago when its build-up started. Hydrogen in the form of H2 and methane probably decreased around 2.4 billion years ago as methane with its hydrogen atoms escaped to the upper atmosphere and space. Methane levels would also be decreased by its easy reaction with dioxygen in the presence of UV light to form CO2. This would paradoxically lead to a cooling of the earth and pronounced glaciation as a more potent greenhouse gas, methane, was replaced with a less potent one, carbon dioxide.
Over the next billion years, dioxygen rose to perhaps 0.2 - 2% (compared to the present levels of 20%) Why? Because the early atmosphere was reducing, the added oxygen combined with a large "sink" of reduced metals (like elemental Cu and Fe) or nonmetals (like C and ammonia), preventing a large buildup. Only after these reduced substances were "titrated" did dioxygen build up to present levels. In addition, the oxygen might have increased weathering (by oxidation) of sulfur deposits which can lead to sulfides entering the ocean, where they could precipitate ocean iron ions that are necessary for cyanobacterial chemistry. This would place constraints on cyanobacterial growth until dioxygen levels in the atmosphere increased enough so sulfides were converted to sulfates. This first increase in atmospheric oxygen is often called the Great Oxidation Event as it correlated and presumably caused one of the greatest mass extinctions (of anaerobic organisms) of all time.
Around 2.3 billion years ago, as trace dioxygen had accumulated in the atmosphere, redox chemistry changed, although isotope evidence suggests that little dioxygen was found in water. Around 1.8 - 1.5 billion years ago, the earth's atmosphere became somewhat oxygenated, which was also coincident with the development of eukaryotic organisms. Until then, life was restricted to the oceans since there was no ozone to absorb dangerous UV radiation. The buildup of dioxygen in the air must have led to another extinction of anaerobic organisms since as we shall see, products of oxygen metabolism are very toxic. Some evolved to use dioxygen. Ozone developed, and life could then migrate from the sea to the land. It wasn't until around 600 million years ago that animals arose, however. Was this event associated with the development of a fully oxygenated (20%) atmosphere? Recent evidence, which shows that substantial oxygen wasn't available in the deep sea until about 600 million years, seems to suggest that. Based on an analysis of iron compounds in waters in Newfoundland, it appears that oxygen was very low in the sea 580 million years ago, during the Gaskier's glaciation period. Immediately after that it rose to levels consistent with atmospheric dioxygen levels of 15%, levels necessary for large animals. Similar trends in carbon and sulfur isotopes in marine rocks in Oman also suggest large increases in oxygen at the end of the Gaskiers glaciation period. What caused this second great oxygenation event? One possibility is that organic matter was sequestered from a reaction with atmospheric dioxygen, as clays bound organic molecules in the ocean and lichens and zooplankton facilitated weather and production of insoluble organic material in the oceans.
Dioxygen is critically important for higher organisms, so an understanding of its chemistry becomes important. This chapter will show that dioxygen is a ground state diradical that has low solubility in an aqueous solution, reacts in a kinetically sluggish fashion in the oxidation reaction, and forms toxic byproducts as it gets reduced. Life forms hence evolved ways to deal with these problems, including ways to increase its solubility (with dioxygen binding and transport proteins), and enzymes (that could activate it kinetically and also detoxify oxygen by-products). Dioxygen is toxic to many cells. Obligate aerobes die in an oxygen environment as many of their cellular components get oxidized by this excellent oxidizing agent. Several strains of bacteria swim away from high levels of dioxygen. A graph showing the log of survival vs log pO2 is linear with a negative slope for a variety of organisms, including mice, fish, rats, rabbits, and insects. Pure oxygen can induce chest soreness, coughs, and sore throats in people. Premature infants put in pure dioxygen environments often developed blindness due to retrolental fibroplasia (a build-up of fibrous tissue behind the lens). The trade-off for this toxicity is clear. Energy is derived from organic molecules through oxidation. Before dioxygen became available to power aerobic catabolism of reduced molecules like fatty acids and less reduced sugars, such molecules were only partially oxidized. The glycolytic pathway, found in most organisms, oxidizes glucose (6 Cs) to two molecules of pyruvate (3 Cs). It was only with the availability of dioxygen did pathways evolve (Kreb Cycle, mitochondrial electron transport/oxidative phosphorylation) that allowed pyruvate to be fully oxidized to carbon dioxide, with the release of much more energy.
The Properties of Dioxygen
It is important to understand the properties of dioxygen since oxidation reactions using its power not only our bodies but our entire civilization. We will concentrate on biological reactions, but even these show the same characteristics as non-biological ones.
- oxidation of organic molecules by oxygen is thermodynamically favored but kinetically slow.
- pure oxygen environments are toxic to cells and organisms.
First, we will try to understand these properties of oxygen, and then we will see how organisms overcome these problems to use dioxygen.
We can understand both of these properties by looking at the molecular orbitals of oxygen and its reduction products as shown in the diagrams below. Ground state oxygen is a diradical, which explains the paramagnetic behavior of oxygen. The two unpaired oxygen each have a spin state of 1/2 for a total resultant spin S of 1, making ground state oxygen a triplet (2S+1) = 3. Organic molecules typically undergo 2 electron oxidation steps. Consider the stepwise oxidation of methane below. The oxidation number of C in methane is -4, -2 in methanol, 0 in formaldehyde, +2 in formic acid, and finally +4 in carbon dioxide, indicating two electron losses in each step. These states are shown in Figure \(\PageIndex{1}\).

The two electrons lost by the organic substrate are added to oxygen, but since the two lost electrons are spin paired, a spin flip must occur to allow the electrons to enter the unfilled oxygen orbitals. Alternatively, energy can be put into ground state dioxygen to produce excited state singlet oxygen (S=0, 2S+1 = 1). The source of the large activation energy required (about 25 kcal or 105 kJ/mol) to flip the electron spin accounts for the kinetic sluggishness of reactions of dioxygen with organic reactants.
A traditional Lewis structure for ground state dioxygen can not be easily written since the electrons are added in pairs, and dioxygen is a diradical. There are 6 electrons in the sigma molecular orbitals from second shell electrons (two each in σ2s, σ2s*, and σ2p,) and 6 electrons in the pi molecular orbitals from second shell electrons (two each in two different π2p orbitals, and one electron each in two different π2p*), so the net number of electrons in bonding orbitals is 4, giving a bond order (or number of 2). In contrast, it is easy to write the Lewis structure of singlet, excited state oxygen, since all electrons can be viewed as paired, with two net bonds (1 sigma, 1 pi) connecting the atoms of oxygen. Figure \(\PageIndex{2}\) shows the molecular orbital diagram for ground and excited state dioxygen.

This Lewis structure will be used to represent singlet, excited oxygen, which should react more quickly with organic molecules. The excited state singlet on the right is unstable and decays to the middle singlet state. The middle state is approximately 94.3 kJ/mol higher in energy than the ground state triplet (on the left). In quantum mechanical parlance, the transition from the ground state triplet to the singlet state is forbidden for several reasons, making it unlikely that absorption of a photon will induce the transition.
The Reduction of Dioxygen
When oxygen oxidizes organic molecules, it is reduced. By adding electrons one at a time to the molecular orbitals of ground-state dioxygen we produce the step-wise reduction products of oxygen. On the addition of one electron, superoxide is formed. A second electron produces peroxide. Two more produce 2 separated oxides since no bonds connect the atoms (the number of electrons in antibonding and bonding orbitals is identical). Each of these species can react with protons to produce species such as HO2, H2O2 (hydrogen peroxide), and H2O. It is the first two reactive reduction products of dioxygen that make it potentially toxic. Figure \(\PageIndex{3}\) shows the MO diagrams for the reduction products of dioxygen.

How are the potential problems in oxygen chemistry dealt with biologically?
Kinetic sluggishness: Enzymes that utilize dioxygen must activate it in some way, which decreases the activation energy. Enzymes that use dioxygen typically are metalloenzymes, and often heme-containing proteins. Since metals such as Fe2+ and Cu2+ are themselves free radicals (i.e. they have unpaired electrons), they react readily with ground-state oxygen which itself is a radical. The molecular orbitals of the metal and oxygen combine to produce new orbitals which for oxygen are more singlet-like. Likewise, dioxygen reacts more readily with organic molecules which can form reasonably stable free radicals, such as flavin adenine dinucleotide (FAD), as we shall see later.
Dioxygen toxicity: Since toxicity arises from the reduction products of oxygen, enzymes that use oxygen have evolved to bind oxygen and its reduction products tightly (through metal-oxygen bonds) so they are not released into the cells where they can cause damage. In addition, enzymes that detoxify free dioxygen reduction products are widely found in nature. For example:
- superoxide dismutase catalyzes the dismutation (self-redox) of 2 superoxides into dioxygen and hydrogen peroxide;
- catalase converts hydrogen peroxide into water and oxygen;
- peroxidase catalyzes the reaction of hydrogen peroxide with an alcohol to form water and an aldehyde
- peroxiredoxins react with peroxides and thioredoxin (a small electron donor) to form water and oxidized thioredoxin.
Finally, free radical scavengers such as vitamins A, C, E, and selenium can react with reactive free radicals to produce more stable free radical derivatives of the vitamins and Se. More on this later.
The Reactions of Dioxygen and its Reduction Products
Triplet O2 - Ground State
Here are some reactions for the ground state (triplet O2):
a. Metals ions - Metal ions are radicals themselves, so can easily react with dioxygen (think about rust). Here is one example
\[\ce{Fe^{2+} + O2 <=> [ Fe^{2+}-O2 <=> Fe^{3+}-O2^{-.}] <=> Fe^{3+} + \underbrace{O2^{-.}}_{superoxide}} \nonumber\]
b. Autoxidation of organic molecules to produce peroxides - These are multistep reactions that have initiation, propagation, and termination steps.
RH → R. (Initiation)
R. + O2 → ROO. (Propagation)
ROO. + RH → R. + ROOH (Propagation)
R. + R. → R-R (Termination)
ROO. + ROO. → ROOR + O2 (Termination)
ROO. + R. → ROOR (Termination
Figure \(\PageIndex{4}\) summarizes the reactions of triplet ground state dioxygen.

The initiation step above occurs mostly at C atoms which can produce the most stable free radicals (allylic, benzylic position, and 3o > 2o >> 10 carbons).
Single O2 - Excited State
Figure \(\PageIndex{5}\) shows some reactions for singlet dioxygen in which dioxygen is shown as with a double bond and two lone pairs on each oxygen.

Alkenes react with oxygen to form hydroperoxides, potentially through an epoxide intermediate. Dienes reacts with oxygen in a Diels-Alder pericyclic reaction to form endoperoxides. A molecular orbital perspective (that you may remember from chemistry classes) on this cycloaddition reaction is shown in Figure \(\PageIndex{6}\).

Singlet oxygen can be made from triplet oxygen by photoexcitation. Alternatively, it can be made from triplet oxygen through collision with an excited molecule which relaxes to the ground state after a radiationless transfer of energy to triplet oxygen to form reactive singlet oxygen. This later process accounts for the photobleaching of colored clothes when the conjugated dye molecules absorb UV and Vis light and relax to the ground state by transferring energy to triplet oxygen to form singlet oxygen. That can more readily react with the conjugated double bonds in the dye. These processes are summarized in Figure \(\PageIndex{7}\).

Superoxide
Common reactions of superoxide are shown below.
- Dismutation: This reaction involves a specified reactant undergoing an oxidation reaction, followed by another molecule of the same reactant undergoing a reduction. \[\ce{O2^{-.} + O2^{-.} + 2H^{+} <=> H2O2 + O2} \tag{slow}\] \[\ce{HO2^{.} + O2^{-.} + H^{+} <=> H2O2 + O2} \tag{fast}\]
- Acid/Base: \[\ce{HO2^{.} <=> O2^{-.} + H^{+}} \tag{pKa = 4.8}\]
- With metal ions: \[\underbrace{\ce{Fe^{3+}}}_{\text{as in heme}} + \ce{O2^{-.}} → \ce{O2} + \ce{Fe^{2+}} \nonumber\]
The enzyme superoxide dismutase catalyzes the dismutation reaction. The common eukaryotic cytosolic form contains Cu2+ and Zn2+ ions, which are coordinated by histidine side chains. The reaction proceeds in two steps or half-reactions. The first is the removal (oxidation) of an electron from superoxide (O2-.) through its reduction by Cu2+. This reaction forms Cu1+ and nontoxic O2.
\[\ce{Cu^{2+}-SOD + O2^{−.} → Cu^{+}-SOD + O2} \nonumber\]
(reduction of Cu; oxidation of superoxide)
The second is the addition (reduction) of an electron from a second superoxide to Cu1+ to reform the catalytic Cu2+ and in the process form the reactive peroxide O22- (unfortunately), which when protonated forms \(\ce{H2O2}\).
\[\ce{Cu^{+}-SOD + O2^{−.} + 2H^{+} → Cu^{2+}-SOD + H2O2} \nonumber\]
(oxidation of copper; reduction of superoxide)
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of the electrostatic potential surface of the superoxidase dismutase dimer showing Cu and Zn ions (2SOD).
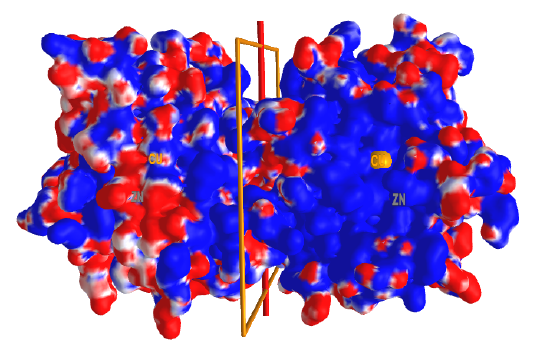
 Figure \(\PageIndex{8}\): Electrostatic potential surface of the superoxidase dismutase dimer showing Cu and Zn ions (2SOD) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zozKPVKr4MzES9
Figure \(\PageIndex{8}\): Electrostatic potential surface of the superoxidase dismutase dimer showing Cu and Zn ions (2SOD) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zozKPVKr4MzES9Two dimers are shown with a rotation C2 axis separating them. Red indicates the negative surface potential and blue the positive. Catalytic Cu2+ and Zn2+ ions are in each subunit and are shown in orange (Cu) and gray (Zn) spheres and labeled (disregard the label not centered on the orange and gray spheres). Note that the Cu2+ and Zn2+ ions are in the center of a large blue surface (positive potential), which helps "sweep up" any negatively charged superoxide O2- nearby. Rotate the complex around the C2 axis and you will see another large positive blue patch on the backside. These two positive electrostatic surfaces facilitate the electrostatic attractions and binding of the dangerous superoxide anion from the larger 3D region around the enzyme.
The Zn2+ in SOD is not redox active. What is its role? Both metals appear to increase the thermostability of the individual monomer and the dimer. In the presence of metal ions (a holo form of the enzyme), the dimer dissociates into monomers at a higher urea concentration than does the apo-dimer. Ion binding reduces the flexibilities of groups found in the dimer interface. The protein has a disulfide bond between Cys 57 and Cys 146. This bond stabilizes a metal ion binding loop that contributes to the binding interface in the dimer. The enzyme operates at diffusion-controlled rates (kcat/KM is between 108 and 109 M-1s-1), in part due to the attraction of any negatively charged superoxide by the electrostatic field around the dimer.
There are two other types of superoxidedismutates, one that uses either Mn2+ or Fe2+ (bacterial, mitochondrial, chloroplasts, protists) and another that uses Ni2+(some prokaryotes).
Peroxide
In contrast to dioxygen which contains multiple bonds between the O atoms, peroxide has only one bond. It is quite weak and requires only 38 kcal/mol (160 kJ/mol) to break it. Remember, bonds can be broken in a heterolytic way (both electrons in a bond go to one of the atoms, or in a homolytic fashion, in which one electron goes to each atom.
Figure \(\PageIndex{9}\) shows typical reactions of peroxides.

The reaction with Fe2+, the Fenton Reaction, is similar to the reaction of triplet O2 with Fe2+. In this reaction, homolytic cleavage of the O-O bond occurs generating OH- and the hydroxy free radical, OH., which will react with any molecule it encounters. Thermal or photochemical homolytic cleavage of peroxide also forms free radicals which react like the hydroxy free radical.
The enzyme catalase facilitates the decomposition of the reactive H2O2 to water and dioxygen. As such offers protection similar to that against superoxide offered by superoxide dismutase. This is the next reaction catalyzed by the enzyme catalase:
The human enzyme is a homotetramer with each monomer having a heme at the active site. The tetramer also binds NADP+ (2/tetramer) but its function is unclear. A potential mechanism for catalase is shown in Figure \(\PageIndex{10}\).

Figure \(\PageIndex{11}\) shows an interactive iCn3D model of human erythrocyte catalase with bound ligand (CN-) and NADPH (1DGG)
%25C2%25A0and_NADPH%25C2%25A0_(1DGG).png?revision=1&size=bestfit&width=312&height=259)
Figure \(\PageIndex{11}\): human catalase with bound ligand (CN-) and NADPH (1DGG). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...TD7yJsfrNcwSJ7
The cyanide ligands in this structure bind to the heme in place of peroxide. The reaction in mammalian catalases appears to involve a tyrosine radical
H2O2 is very similar in structure to H2O. How does the enzyme differentiate between them? Both approach the heme through a long 25 Å water channel with a hydrophobic constriction part way into the channel leading to the active site heme. Four waters in the crystal structure are positioned in the constricted opening and in a widened open just at the heme. The amino acid side chains forming the constriction are Val 74, Val 116, Pro 129, Phe 153, Phe154 and Trp 186. These allow only small molecules to enter. Two of the four waters form hydrogen bonds to side chains (one to His 75 and Asn 148 and another to Gln 168 and Asp12. The others don't form hydrogen bonds, are more dynamic, and more likely to leave the channel. H2O2 is bigger and can form bridging hydrogen bonds to side chains that the mobile waters can't. They probably leave the opening. H2O2 is more polar and has a higher dipole moment (2.26 Debye) compared to water (1.86 Debye) which implies that it would be differentially stabilized by hydrogen bonds in this hydrophobic site compared to the less polar water.
Figure \(\PageIndex{12}\) shows an interactive iCn3D model of human erythrocyte catalase (monomer) showing the hydrophobic constricture leading to the active site (1DGF)
.png?revision=1)
Figure \(\PageIndex{12}\): Human erythrocyte catalase (monomer) showing hydrophobic constriction leading to the active site (1DGF). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...e48pAa9X4zhcM7
Hydroxyl Free Radical
We won't specifically discuss the reaction of the hydroxyl free radical (.OH-) since it will react with anything nearby to produce another free radical. General reactions of the radical are shown in Figure \(\PageIndex{13}\).

In summary, we reap the benefits of using dioxygen as an oxidizing agent as it allows the aerobic and hence complete oxidation of carbohydrates and lipids to CO2 and H2O. Yet we pay the price for using O2 as an oxidizing agent as its partial reduction products, the superoxide radical, peroxides, and hydroxy free radical, collectively known as reactive oxygen species (ROS) can react with and damage proteins, nucleic acids, and lipids, as we will see below. Figure \(\PageIndex{13}\) shows a summary of their reactions.

We have presented the role of superoxide dismutase and catalase in the removal of ROS in cells. In addition to these elegantly designed proteins, there is another simpler biomolecule, the tripeptide glutathione (γGlu-Cys-Gly), that can reduce ROS in cells and prevent oxidative damage. Glutathione serves as a major antioxidant in cells. It exists in reduced (GSH) and oxidized (GSSG) states, the ratio of which depends on the cellular oxidative stress. It can react with and detoxify peroxides and alkyl free radicals by the following net reactions:
2 GSH + R2O2 → GSSG + 2 ROH
2GSH + 2R. → GSSG + 2RH
Protection from Fe2+ - Ferritin and Transferrin
The Fenton reaction shows the potential problem with having free Fe2+ ions in a chemical state that would easily allow this reaction and the generation of ROS. Hence much of the Fe2+ in the body is sequestered in Fe2+ binding cofactors like heme and FeS clusters. It is transported in the blood by the protein transferrin and stored in cells like the erythrocyte in ferritin. The iron ions in both transferrin and ferritin are in the +3 oxidation state (Fe3+). This strongly positive cation is quite insoluble in the presence of anions like hydroxide, phosphate, and carbonate. The Ksp values for the hydroxide salt of Fe3+ is 3.8x10-38. Let's detour for a bit and look at the structures of ferritin and transferrin and how they work, starting with ferritin.
Ferritin
The biologically functional form of ferritin is a 24-mer. The structure encapsulates a large volume that can hold many Fe ions (up to 4500) in the central cavity. The ions are stored in the more insoluble form, Fe3+, in complexes of oxide and hydroxide. Mammalian ferritin contains both heavy (H) and light (L) chains so they are hetero 24-mers.
Figure \(\PageIndex{15}\) shows an interactive iCn3D model of ferritin, the intracellular Fe storage protein (1fha).
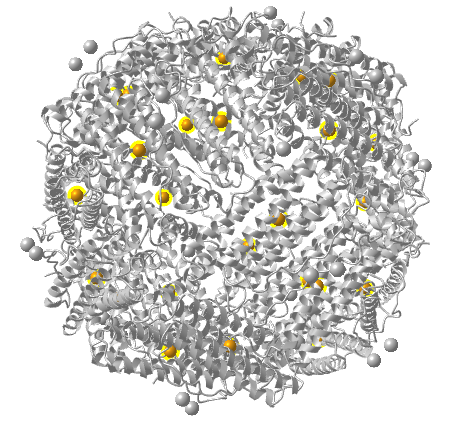
Figure \(\PageIndex{15}\): Ferritin, the intracellular Fe storage protein (1fha). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...MQXAbPAwNnVya8
The ferritin chains are shown in purple. This structure shows only 24 Fe ions (in orange spheres with yellow halo).
Two questions arise. How does Fe2+ get into the internal volume of ferritin and how does it get converted to Fe3+? There must be a channel through which Fe2+ can diffuse. Its conversion to Fe3+ requires catalysis by an Fe cluster in the heavy chain (H) subunits. Figure \(\PageIndex{16}\)s shows a generic diagram outlining the processes of uptake and conversion to Fe3+ salts inside the central cavity of ferritin.

Heart and brain ferritin are enriched in the heavy (H) chain. These two organs clearly require safety from toxic Fe2+ ions. Ferritins in organs like the liver and spleen, which store lots of iron, are enriched in the light (L) chain. The two chains are about 50% homologous, but the H chain has a dinuclear ferroxidase iron site which catalyzes the Fe2+ to Fe3+ conversion. Once inside the L chain surface provides a nucleation site for the deposition of Fe3+ into a ferrihydrite "precipitate" ((Fe3+)2O3•0.5H2O). The general reaction is:
2Fe2+ + O2 → [Fe3+-O-O-Fe3+] → [Fe3+-O(H)-Fe3+]
Figure \(\PageIndex{17}\) shows a possible generic mechanism for the oxidation of the Fe cluster from Fe2+ to Fe3+.

It acts as a ferroxidase that suggests that dioxygen is involved as a ligand in the oxidation of the two Fe2+ ions in the cluster to Fe3+.
Figure \(\PageIndex{18}\)s shows a closeup of the interactions of the di-Fe cluster and two other Fe ions bound in the human H chain with water ligands.
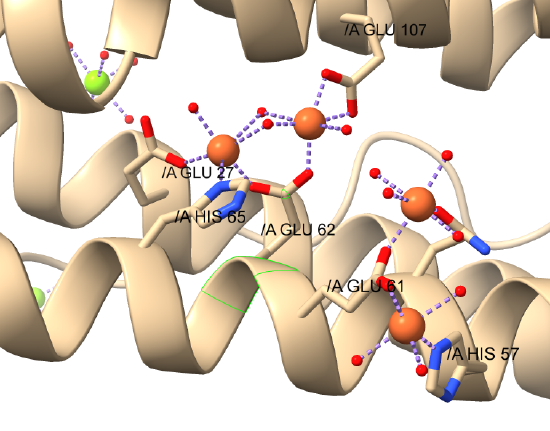
The dinuclear Fe core is shown in the central area of the figure.
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of human heavy-chain ferritin monomer with bound Fe (4zjk)
.png?revision=1)
Figure \(\PageIndex{19}\): Human heavy-chain ferritin monomer with bound Fe (4zjk). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?Ux6ED11dT5dyBb2XA
The crystal structure of the full ferritin structure shows multiple binding sites and a channel to the oxidase site.
Now let's look at how the light chain L might nucleate the formation of the iron precipitates. Crystal structures show how mineralization probably occurs at a specific site on the light chains that present themselves on the inside surface of ferritin. Figure \(\PageIndex{20}\) shows a closeup of the interactions of a di-Fe cluster and two other Fe ions bound in the human L chain with water and peroxide ligands. This site probably represents the nucleation and mineralization site.
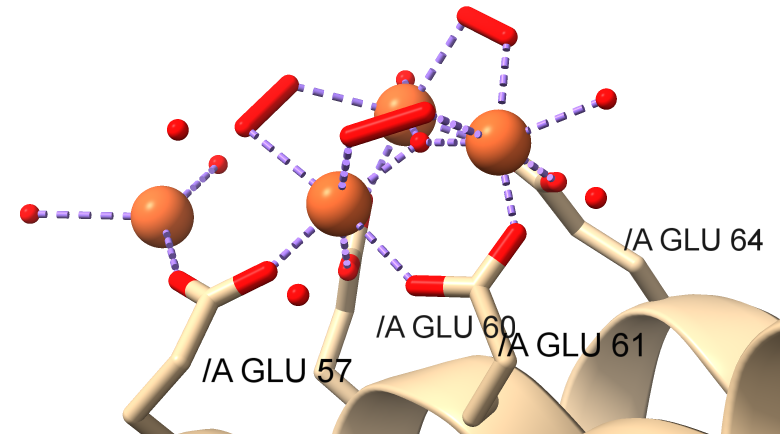
Figure \(\PageIndex{21}\) shows an interactive iCn3D model of human light chain ferritin with a possible nuclear site for mineralization (5LG8). The structure was made after 60 minutes of mineralization.
.png?revision=1)
Figure \(\PageIndex{21}\): Human human light chain ferritin with a possible nuclear site for mineralization (5LG8). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...LKo7WJtoHeLBq9
The structure suggests the presence of a μ(3)-oxo)Tris[( μ (2)-peroxo)] triiron(III) cluster assembled at subsite on the L chains containing the carboxylate ligands Glu60, Glu61, and Glu64 side chains. A Glu57, which is along the incoming path of Fe ions, is involved in Fe delivery and coordination. Figure \(\PageIndex{22}\) shows the electrostatic surface around the nucleation site.
ElectroSurface.png?revision=1&size=bestfit&width=292&height=291)
Note that the Fe ions are embedded in a site of negative electrostatic potential arising, in part, from the localization of the glutamic acid side chains in the site. this
Why doesn't the heavy chain of ferritin perform the same nucleation function in preparation for the crystallization of ferrihydrite? A comparison of Figures 18 and 20 shows that the H chain (which has the ferroxidase activity) has a His 65 ligand instead of a glutamic acid (position 60) as one of the coordinating ligands. This gives Glu 61 more flexibility which must inhibit the nucleation and mineralization process.
Figure \(\PageIndex{23}\) shows two top views (left and center image) and one side view of three contiguous subunits of human heavy-chain ferritin monomer with bound Fe (4zjk). The gray/black sphere is actually a Ca2+ ion (which is larger than Fe2+) from the crystal structure. This three-monomer cluster would be replicated 8 times to form the full ferritin shell. There is a three-fold C3 axis going through the central calcium in the figure where all the monomers meet. Fe2+ must move through these central ions into the internal cavity.

Transferrin
Iron ions are moved in the circulation bound to the iron-binding protein transferrin. It binds to a transferrin receptor and can be endocytosed into the cell, where the Fe ions are transferred and stored in ferritin. The transferrin receptor can also bind and internalize circulating ferritin (see pdb 6GSR).
Figure \(\PageIndex{24}\) shows an interactive iCn3D model of transferrin (2N and 2C-lobes) binding to the ectodomain of the transferrin receptor (1SUV).
..png?revision=1&size=bestfit&width=374&height=335)
Figure \(\PageIndex{24}\): Transferrin (2N and 2C-lobes) binding to the ectodomain of the transferrin receptor (1SUV). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...cBhTyxoSDQgH47
The transferrin receptor is shown in gray (2 monomers) bound to the N-lobes (cyan) and C-lobes (magenta) of transferrin. Each transfer lobe has a bound CO32- (spacefill with CPK colors) and a Fe3+ ion (orange).
Oxidative Modification of Proteins
Many amino acid side chains can be oxidized in cells, as shown in Table \(\PageIndex{1}\).
| Amino acids | Oxidation products |
|---|---|
| Cysteine | Disulfides, cysteic acid |
| Methionine | Methionine sulfoxide, methionine sulfone |
| Tryptophan | 2-, 4-, 5-, 6-, and 7-Hydroxytryptophan, nitrotryptophan, kynurenine, 3-hydroxykynurinine, formylkynurinine |
| Phenylalanine | 2,3-Dihydroxyphenylalanine, 2-, 3-, and 4-hydroxyphenylalanine |
| Tyrosine | 3,4-Dihydroxyphenylalanine, tyrosine-tyrosine cross-linkages, Tyr-O-Tyr, cross-linked nitrotyrosine |
| Histidine | 2-Oxohistidine, asparagine, aspartic acid |
| Arginine | Glutamic semialdehyde |
| Lysine | α-Aminoadipic semialdehyde |
| Proline | 2-Pyrrolidone, 4- and 5-hydroxyproline pyroglutamic acid, glutamic semialdehyde |
| Threonine | 2-Amino-3-ketobutyric acid |
| Glutamyl | Oxalic acid, pyruvic acid |
We will focus just on one here, the oxidation of the ε-amino group of lysine by H2O2 to form α-aminoadipic semialdehyde, where the amine is replaced with an aldehyde. This reaction is called the Fenton reaction as is actually a carbonylation reaction. A hypothetical mechanism for the oxidation of lysine side chains is shown in Figure \(\PageIndex{25}\).

The next result is the oxidation of the lysine ε-amino group to an aldehyde. Oxidized levels of proteins (as evidenced by increased levels of carbonylation) increase dramatically with age (especially after age 40 ). The reactions seem to be catalyzed by metals and may proceed by the generation of hydroxy free radicals. Diseases associated with premature aging (Werner's Syndrome, another link to Werner's Syndrome, Progeria) show very high levels of oxidized proteins at an early age. Fibroblasts from 10 yr. old children with progeria have levels of oxidized proteins usually not seen until the age of 70. Beta-amyloid protein deposits (found in Alzheimer's and Down's Syndrome) cause neurotoxicity and death, partly by increasing superoxide production by endothelial cells, causing vasoconstriction/dilation, and ultimately disease progression. Beta-amyloid aggregates appear to increase H2O2 levels, in a process facilitated by Fe2+ and Cu+. Free radical scavengers (antioxidants) may help to prevent this damage.
Carbonylation of proteins appears to be irreversible and nonrepairable. Increased carbonylation leads to misfolding and protein aggregation in ways in which protein chaperones can not reverse. The graphs in Figure \(\PageIndex{26}\) (the summation of many experiments) show the correlation (negative) of increasing carbonylation of proteins (red line), a measurement of oxidative damage, and the resulting decrease in protein function (green).

Red dots in the top representations of protein show carbonylation. Variants of the same protein (proteoforms) that have more intrinsically disordered regions (m1) are more susceptible to carbonylation compared to more ordered variants (m3). Cancer starts to increase around 40 years of age, and the levels of carbonylation correlate with increased cancer rates and may, in part, cause it.
Lou Gehrigs Disease (Amyotrophic Lateral Sclerosis) is a disease of progressive motor neuron degeneration, which affects 1/100,000 people, and is 10-15% familial. Of the familial cases, about 25% have a mutation in superoxide dismutase I, a copper-zinc enzyme. About 2-3% of ALS patients carry 1 of 60 different dominant mutations in this enzyme. Mutations often decrease the stability of the protein which decreases Zn2+ affinity 5-50 fold. The A4V mutation (valine at amino acid 4 substituted for Ala) has the weakest Zn affinity and causes rapid disease progression. In the absence of Zn2+, the apoprotein somehow seems to induce cell death in neurons. This superoxide dismutase also expresses a second activity. It also acts as a peroxidase which takes ROH + H2O2 to form an RHO (an aldehyde) plus water. In some cases, the enzyme retains normal activity against superoxide but altered peroxidase activity.
Figure \(\PageIndex{27}\)s shows the extent of carbonylation of wild type (WT) and two mutant forms α-synuclein after exposure (in vitro) to increasing doses of γ-radiation. α-synuclein forms aggregates (Lewy bodies) in Parkinson's Disease.

The mutants, especially the A53T one, show significantly high extents of oxidative damage. This particular mutation is associated with the early onset (around 30) of Parkinson's Disease.
Is your hair going white?: Wood et al have shown that millimolar concentrations of hydrogen peroxide builds up in hairs that have grayed and whitened. This was associated with a decrease in catalase and in increases in Met oxidation (to Met-sulfoxide) in proteins, also associated with a decrease in the repair enzyme Met-sulfoxide reductase, Met 374 in the active site of tyrosinase, an enzyme required for the production of melanin in hair follicles, is also damaged, leading to lack of melanin, a pigment necessary for hair coloration and "senile hair graying".
ROS and Protein Folding
As discussed earlier, the cytoplasm has sufficient concentrations of "β-mercaptoethanol"-like molecules (used to reduce disulfide bonds in proteins in vitro) such as glutathione (γ-Glu-Cys-Gly) and reduced thioredoxin (with an active site Cys) to prevent disulfide bond formation in cytoplasmic proteins. Disulfide bonds in proteins are typically found in extracellular proteins, where they serve to keep multisubunit proteins together as they become diluted in the extracellular milieu. These proteins destined for secretion are cotranslationally inserted into the endoplasmic reticulum (see below) which presents an oxidizing environment to the folding protein and where sugars are covalently attached to the folding protein and disulfide bonds are formed (see Chapter 3D: Glycoproteins - Biosynthesis and Function). Protein enzymes involved in disulfide bond formation contain free Cys which form mixed disulfides with their target substrate proteins. The enzymes (thiol-disulfide oxidoreductases, protein disulfide isomerases) have a Cys-XY-Cys motif and can promote disulfide bond formation or their reduction to free sulfhydryls. They are especially redox-sensitive since their Cys side chains must cycle between and free disulfide forms.
Reactive oxygen species (ROS) can significantly affect redox chemistry, and if present in excess can place the cell in a condition of "oxidative" stress. ROS can indiscriminately oxidize lipids, nucleic acids, and proteins, but more specifically, they may also oxidize proteins involved in creating and maintaining the normal disulfide bond formation in proteins. As the concentration of ROS increases, the concentration of cytoplasmic proteins with incorrect disulfides should increase. Using a two dimension PAGE system (first dimension run under nonreducing and the second reducing conditions) of neural cell proteins derived from cells exposed to normal and differing oxidative conditions (hydrogen peroxide or decreased intracellular glutathione levels, Cumming et al showed that oxidizing stress increased the levels of disulfide bonds in redox sensitive enzymes and, unexpectedly, among other cytoplasmic proteins involved in many aspects of life, affecting the activity of many cellular processes, suggesting that disulfide bond formation may have not only a structural but regulatory role.
Oxidative Modification of Lipids:
Figure 4 shows the most likely position in organic molecules that can form stable free radicals (allylic, benzylic position, and 3o > 2o >> 10 carbons) are likely targets for reaction with ROS. Hence unsaturated fatty acids are extra reactive at the methylene C that separates the double bonds as shown in Figure \(\PageIndex{28}\).

Lipid and protein oxidation - cardiovascular disease
The initial stages of cardiovascular disease appear to involve the development of fatty acid streaks under the artery walls. Macrophages are immune cells that have receptors that recognize oxidized lipoproteins in the blood, which they takeup. The cells then further differentiate into fat-containing foam cells which form the streaks. Oxidation of fatty acids in lipoproteins could produce lipid peroxides and along with the Fenton reaction lead to the oxidation of apoproteins in LDL. Cortical neurons from fetal Down's Syndrome patients show 3-4 times levels of intracellular reactive oxygen species and increased levels of lipid peroxidation compared to control neurons. This damage is prevented by treatment of the neurons in culture with free radical scavengers or catalase. Key events in atherosclerotic plaque initiation are shown in Figure \(\PageIndex{29}\).

How do fatty streaks appear under the endothelial cells? LDL oxidized in the lipid monolayer and through carbonylation of lysine side chains of the apoproteins in LDL binds to "scavenger" receptors in macrophages which have moved into the intima below the endothelial cell barrier. Scavenger receptors were first recognized to bind acetylated LDL (conversion of ε-amino groups of lysines, for example, to acetylated and uncharged derivatives). This mimics to some degree the carbonylation of the ε-amino groups to aldehydes, as shown in Figure \(\PageIndex{30}\).

Either modification would make the apoprotein more acidic with a lower isoelectric point since positive lysine side chains are replaced with neutral derivatives. Assuming an asymmetric distribution of the negatively charged side chains (Asp and Glu) on the apoprotein, any pre-modification negative electrostatic potential surfaces on the apoprotein would become more negative, enhancing binding to positive clusters displaying positive electrostatic potentials on scavenger receptors. Scavenger receptors often bind polyanions.
There are many 12 different classes (A-L) of scavenger receptor classes that have been identified. One, class C, is only found in drosophila. They bind a variety of polyanionic ligands and display broad binding specificity. Many in a single class have multiple names that makes their designation even more confusing. They bind a diverse set of ligands including those from bacteria and yeast (in a way similar to pathogen-associated molecular patterns - PAMPs - in the innate immune system) as well as self and modified self ligands (such as oxidized LDL - oxLDL). Once bound the ligand and scavenger receptors are taken into the cell by endocytosis for removal and degradation of the bound ligand. They can also act in signaling pathways.
The members of the scavenger receptor family are designated as illustrated in this example, SR-F1.1, where S is Scavenger, R is Receptor, F is Class, 1 is Order in class and 1 is alternatively spliced forms. Figure \(\PageIndex{31}\) shows domain structures of the different classes of scavenger receptors.
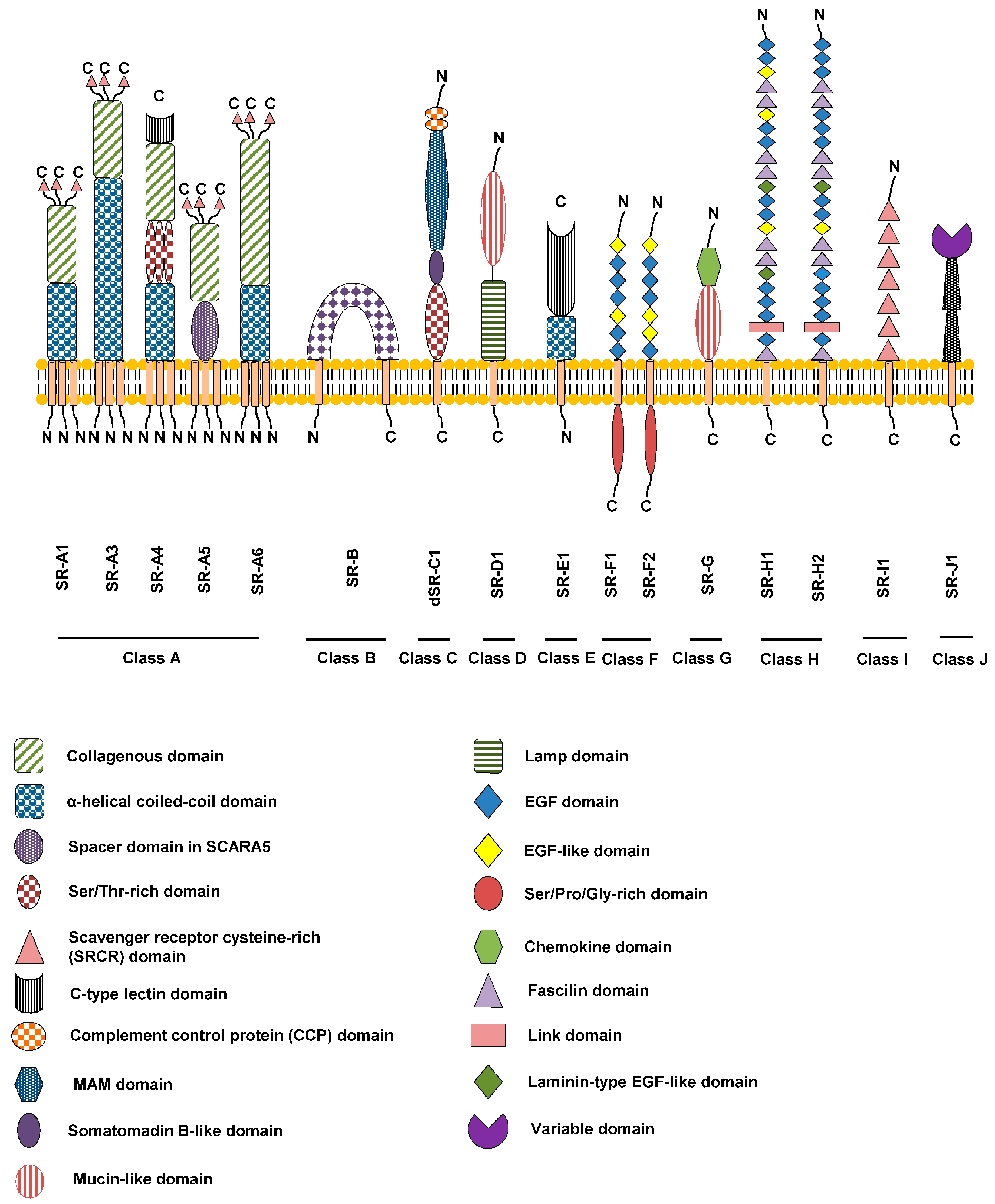
Figure \(\PageIndex{31}\): Domain structures of the different classes of scavenger receptors. Zani et al. Cells 2015, 4, 178-201. https://doi.org/10.3390/cells4020178. Creative Commons Attribution 4.0 International
A more detailed cartoon showing examples from a few different scavenger receptor classes involved in cardiovascular disease is shown in Figure \(\PageIndex{32}\).

Modified LDL binds to several different scavenger receptors, including SR-A1 (also called SCARA1 or CD204), SR-A2 (also called MARCO), and SR-E1 (also called Lectin-like oxidized LDL receptor 1 or LOX-1) binds oxidized and acetylated LDL.
Let's look in greater detail at two scavenger receptors that recognized oxLDL
SR-A2 (MARCO)
This scavenger receptor is a trimer that binds oxLDL, polyanions, and pathogens. It has an extracellular domain (ectodomain) formed from three monomers that are cysteine rich, so it's abbreviated SRCR. A five-stranded antiparallel β-sheet, and an α-helix with a large loop covering it, while the dimer has a larger 8-stranded eight-stranded β-sheet. The polyanion ligands bind presumably to the surface of the receptors with a positive (blue in figures) electrostatic potential associated with an arginine cluster. Crystal structures show that the protein also has a region of negative (red in figures) electrostatic potential which most likely is involved in metal ion binding and in the self-association of monomers to form trimeric receptors.
Given the size of the oxidized LDL (250 Å in diameter), it would not be unexpected that oxLDL binding would promote the formation of clusters of the normally trimer scavenger receptor. This is illustrated in Figure \(\PageIndex{33}\).

Monomeric, dimeric, and oligomeric forms of SR-A2 (MARCO) are shown in the bottom part of the figure. The trimeric receptor molecules can form dimers and multimers by swapping domains. Multiple interactions would promote tighter binding of large ligands such as LDL and even bacteria (0.2-2 μm diameter). The assembly could proceed to the formation of oligomers (the yellow molecule has swapped domains with three other molecules), thus resulting in the creation of a large surface capable of interacting with large ligands, such as modified LDL (250 Å in diameter) or bacteria (0.2-2 μm). The red and blue surfaces shown above the trimer represent the negative (red) and positive (blue) electrostatic surface electrostatic potential of the oligomeric from top down.
SR-E1 (Lox1 or Lectin-like oxidized LDLR or Oxidized low-density lipoprotein receptor 1.
LOX-1 is expressed on macrophages, dendritic cells, endothelial cells, platelets, smooth muscle cells, and adipocytes. It binds oxLDL, some bacteria (through their negatively charged cell walls), and even apoptotic cells. Figure \(\PageIndex{34}\) shows an interactive iCn3D model of the extracellular C-type lectin-like domain of dimeric human Lox-1 (1YPQ)
.png?revision=1&size=bestfit&width=385&height=263)
Figure \(\PageIndex{34}\): Extracellular C-type lectin-like domain of dimeric human Lox-1 (1YPQ). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...qGY4HBzPjjs7P7
The dimer in this structure is connected by a disulfide bridge. The two monomers are shown in gray spheres. They come together to form a heart-like structure A series of arginines (with blue spheres for the surface Ns and labeled) are shown in the cradle of the heart shape. These most likely interact with the oxidized apoprotein B of the oxLDL. Other blue (N) and red (O) spheres near to each other can form salt bridges and may interact with zwitterion heads of LDL surface lipids such as phosphatidylcholine, sphingomyelin, or phosphatidylethanolamine.
Figure \(\PageIndex{35}\) shows an interactive iCn3D model of an AlphaFold predicted model of human oxidized LDL receptor - LOX (P78380)
.png?revision=1&size=bestfit&width=142&height=272)
Figure \(\PageIndex{35}\): . (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...rLoi7ZK4K3SsH8
The yellow spacefill indicates the transmembrane segment. Rotate the model along the long axis and you will see one face of the top domain has a red (negative electrostatic potential) face while the opposite side has blue (positive potential).
Oxidative Modification of DNA
Significant evidence suggests oxygen free radicals are linked to aging and diseases. Mutations caused by hydroxylation reactions (presumably from the generation of hydroxyl free radicals as shown above) can potentially lead to cancer. A particularly nasty reaction is the insertion of the hydroxy radical into bases in DNA. Figure \(\PageIndex{36}\) shows the hydroxylation at position 8 of guanine to produce 8-oxy-G and at positions 5 and 6 in thymine.

Mitochondrial DNA is more susceptible to oxidation than is nuclear DNA. The human mitochondrium has a small genome (16.5 Kb compared to the nuclear genome of 3 Gb) which code 13 protein subunits involved in respiration, 22 tRNAs and two ribosomal RNAs. (The mitochondria presumably are vestiges of a bacteria which invaded an early cell and established a symbiotic relationship with the cell). There is an inverse correlation of oxidized mitochondrial DNA [8-oxoG] with the maximal life span of an organism, but this correlation is not seen with nuclear DNA. Presumably, the nuclear DNA is somewhat protected from oxidative damage since it is bound to histone proteins (which form nucleosome core particles with DNA) and by DNA repair enzymes. DNA repair enzymes that are encoded in the nucleus are found in the mitochondria and mitochondrial DNA is packaged with mitochondrial transcription factor A (TFAM). Examination of human bladder, head and neck, and lung primary tumors reveals a high frequency of mitochondrial DNA mutations. In addition, most dioxygen use by the cell occurs in the mitochondria. Hence this organelle probably faces the highest concentration of toxic oxygen reduction products. Recently, the crystal structure of an enzyme, adenine DNA glycosylase (MutY), that repairs 8-oxyG modified DNA has been determined in complex with the oxidatively damaged DNA. If not repaired, the 8-oxyG base pairs with adenine instead of cytosine, causing a GC to AT mutation on DNA replication.
Figure \(\PageIndex{37}\) shows an interactive iCn3D model of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase (1RRQ)
.png?revision=1&size=bestfit&width=431&height=312)
Figure \(\PageIndex{37}\): Adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase (1RRQ) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?4RiEc6TuqmexiW1r7The protein MutY, which catalyzes the base excision and repair, is shown in gray. The DNA strands are shown in magenta and blue. 8-OxyG on the magenta strand is labeled 8OG7 and is shown in CPK-colored sticks. Its mismatched adenine base pair partner, labeled A18 on the blue strand, is shown in CPK-colored sticks. Notice its orientation is kinked away from the orientation in a canonical base pair. Key amino acid side chains (Thr49, Leu86, Tyr88, and Ser308) interacting with the 8-oxyG are shown in CPK-colored sticks and labeled.
Although oxidative damage in mitochondria clearly can promote premature aging, other independent mechanisms may also. Kujoth et al. developed a mouse model that expressed a mutant form of mitochondrial DNA polymerase that was defective in the proofreading activity of the enzyme. These mice displayed premature aging but showed no increased levels of oxidized mitochondrial lipids or hydroxylated G residues in mitochondrial DNA. They did show significant activation of a cytosolic enzyme called caspase-3, which when active lead to the programmed death of cells (a process called apoptosis). This calcium-activated aspartic acid protease (with an active site Asp) is activated by binding mitochondrial cytochrome C that has "leaked" into the cytoplasm from its normal location in the intermembrane space in mitochondria. The process is usually associated with DNA damage (mutations, fragmentation) that would arise if the proofreading function of DNA polymerase was defective. This was indeed found in these mice.
Oxidative damage to biomolecules might not initiate aging and disease processes, but rather might be a secondary effect of other initiating events. .Reversing or preventing oxidative damage might slow the progression of aging and disease. Aging is a complex feature of organisms and would be expected to have complex causes and biological effects. At the organismal level, aging has been studied in the roundworm C. elegans which lives for only a few weeks. Genetic analyses can be easily used to find gene alterations associated with premature aging. One hormonal system that has recently been associated with aging in eukaryotes (and in C. elegans) involves the signaling pathways for insulin and insulin growth factor I (IGF-1), which regulate carbohydrate, lipid, and reproductive pathways in C. elegans. Mutations that decrease signaling from this pathway increase C. elegans life span. These mutations lead to increased activity of the DAF16 transcription factor, which upregulates the expression of many genes. In contrast, wild-type organisms, when exposed to insulin or IGF-1, decrease the activity of DAF16. Using DNA microarrays, investigators determined which DAF16-controlled genes were upregulated in mutant worms in the mid-life point of the organism. These genes included, among others, peroxisomal and cytosolic catalase, Mn-superoxide dismutase, cytochrome P450s, metallothionein-related Cd-binding protein, and heat shock proteins. We will investigate the function of several of these gene products in the next section, but needless to say, they are all involved in cellular responses to stress, often involving dioxygen metabolites. The over-expression of mitochondrial catalase in mice increased their lifespan by 20%. It has also been showed that decreased levels of insulin-like growth factor also promote longevity in mice, indicating again that mechanisms in addition to oxidative damage by ROS are involved in aging.
Beneficial Oxidation of Proteins: Oxidative Burst in Macrophages
There are cases in which oxidative damage to protein and lipids is desirable. One example involves the role of macrophages in the immune system in eliminating foreign microorganisms. When macrophages recognize and engulf microbes, one mechanism deployed in killing the microorganism is through oxidative damage. The stimulated macrophages undergo an oxidative burst which leads to increased oxygen utilization. One outcome of this is the generation of ROS. The activation of the ROS-generating system can also kill the macrophage (which is OK).
In addition, immune function decreases with age. This probably also occurs through damage from ROS. Telomeres at the end of chromosomes are also shortened by oxidative stress and irradiation. They are enriched in guanine bases the have may repeat (thousands) of TTAGGG sequences and hence are susceptible to oxidation. However, as you might expect, macrophages have developed ways to limited self-damage by ROS.
In the presence of ROS, the macrophage or mitochondrial kinase Mst1/2 are recruited to their respective membranes from the cytosol. The enzyme acts as a ROS sensor and "attenuator" though the phosphorylation and stabilization of a protein Keap1 that binds to a transcription factor Nrf2 (also called NFE2L2), a transcription factor. When bound to unphosphorylated Keap1, the transcription factor Nrf2 is target for proteolysis. Keap1 phosphorylation prevents its binding to Nrf2. Free Nrf2 then can translocate to the nucleus where it promotes transcription of antioxidant proteins such as glutamate-cysteine ligase catalytic subunit (Gclc), which catalyzes the first step and rate-limiting step of glutathione (g-glutamyl-cysteinyl-glycine) synthesis. These processes are illustrated in Figure \(\PageIndex{38}\).

Phagosomal or mitochondrial ROS release attracts Mst1/2 to the membrane of phagosome or mitochondrion from the cytosol and activates Mst1/2; Mst1/2
phosphorylate Keap1 to stabilize Nrf2 and regulate the expression of antioxidant enzymes to protect the cell against oxidative damage.