
6.8: Cofactors and Catalysis - A Little Help From My Friends
- Page ID
- 21161


Cofactors and Electron Pushing: Sources and Sinks
To make and break bonds, electrons have to be moved. In drawing reaction mechanisms, we showed how electrons move from "sources" to "sinks". In many enzyme-catalyzed reactions, vitamin derivatives are used as substrates or "cofactors" or "coenzymes" to facilitate the flow of electrons in bond-making and breaking. The section focuses on cofactors, which facilitate the flow of electrons from the substrate to product. We will see these enzymes in more detail in specific chapter sections.
Cofactors are molecules that bind to enzymes and are required for catalytic activity. They can be divided into two major categories: metals and coenzymes. Metal cofactors commonly found in human enzymes include iron, magnesium, manganese, cobalt, copper, zinc, and molybdenum. Coenzymes are small organic molecules that are often derived from vitamins. Coenzymes can bind loosely with the enzyme and release from the active site. As such, they are also considered substrates for the reaction. Alternatively, they may be tight binding and cannot dissociate easily from the enzyme. In this case, after their initial participation in an enzyme-catalyzed reaction, the enzyme would no longer be able to use the cofactor in another round of catalysis until the initial state of the cofactor is reformed, which takes another chemical reaction and often an additional substrate.
Tight-binding coenzymes are referred to as prosthetic groups. Enzymes not yet associated with a required cofactor are called apoenzymes, whereas enzymes bound with their required cofactors are called holoenzymes. Sometimes organic molecules and metals combine to form coenzymes, such as in the case of the heme cofactor (Figure 7.15). Coordination of heme cofactors with their enzyme counterparts often involves interactions with histidine residues, as shown in the succinate dehydrogenase enzyme shown in Figure \(\PageIndex{1}\).
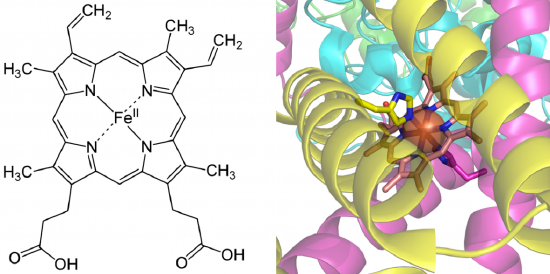
Many biological cofactors are vitamin B derivatives, as shown in Table \(\PageIndex{1}\) below. Many vitamin deficiencies cause disease states due to the inactivity of apoenzymes that can not function without the correctly bound coenzyme.

Table \(\PageIndex{1}\): Essential B-Vitamins and their Modified Enzyme Cofactors
Cofactors can help to mediate enzymatic reactions through the use of any of the different catalytic strategies listed above. They can serve as nucleophiles, mediate covalent catalysis, form electrostatic interactions with the substrate, and stabilize the transition state. They can also cause strain distortion or facilitate acid-base catalysis. Metal-aided catalysis can often use homolytic reaction mechanisms that involve radical intermediates. This can be important in reactions such as those occurring in the electron transport chain that requires the safe movement of single electrons.
We present plausible mechanisms for prototypical reactions using some of the cofactors shown in Table \(\PageIndex{1}\) above. Each shows the flow of electrons from a source to a sink. The source is often a pair of electrons on an anion, formed by the prior removal of a proton from the atom by a general base. A sink could be a carbonyl O, which receives a pair of electrons from one of the C=O bonds of the carbonyl. As a bond is made to the carbonyl, one of the double bonds must break with the electrons going (temporarily if the reaction is a nucleophilic substitution reaction) to the carbonyl O, an excellent sink since it is so electronegative. An even better sink is a positive N of an iminium ion; examples are shown below. Just the "business parts" of the cofactors are shown below.
To appreciate the mechanism used by cofactors, and show a clear example of an electron source/sink, let's look at a reaction that doesn't require a cofactor, the spontaneous decarboxylation of a β-keto acid as shown in Figure \(\PageIndex{1}\).

Even though no cofactor is required, nucleophilic catalysis by an amine through Schiff Base formation would speed up the reaction (as we will see below). Now let's look at how some of the cofactors listed in Table \(\PageIndex{1}\) above facilitate electron flow in reactions.
Thiamine pyrophosphate - decarboxylation of α-keto acids
Thiamine pyrophosphate (TPP) facilitates the decarboxylation of α-keto acids. TPP is a derivative of thiamine, vitamin B1, whose deficiency causes beriberi. TPP is covalently attached to the enzyme, such as in pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, two enzymes that catalyze the decarboxylation of α-keto acids. The structure and "business" end of TPP and its catalytic activity are shown in Figure \(\PageIndex{2}\).

The number of arrows leading to the product does not reflect the actual number of steps.
Figure \(\PageIndex{3}\) shows an interactive iCn3D model of the thiamin diphosphate-dependent enzyme pyruvate decarboxylase from the yeast Saccharomyces cerevisiae (1pvd).
.png?revision=1&size=bestfit&width=396&height=335)
Flavin Adenine Dinucleotide (FAD) - hydride transfer
FAD and its reduced form, FADH2, are tightly or covalently attached to an enzyme, so FAD must be regenerated in each catalytic cycle. Figure \(\PageIndex{4}\) shows an example of how this cofactor facilitates the transfer of a :H- hydride ion to the "business end" of FAD. In contrast to a transfer of protons (H+), an acid/base reaction, hydride transfer removes 2 electrons from the substrate (in this case, succinate) along with a proton in an oxidation reaction as FAD is reduced.

Figure \(\PageIndex{5}\) shows an interactive iCn3D model of the FAD-binding domain of cytochrome P450 BM3 from Priestia megaterium in complex with NADP+ (4DQL)
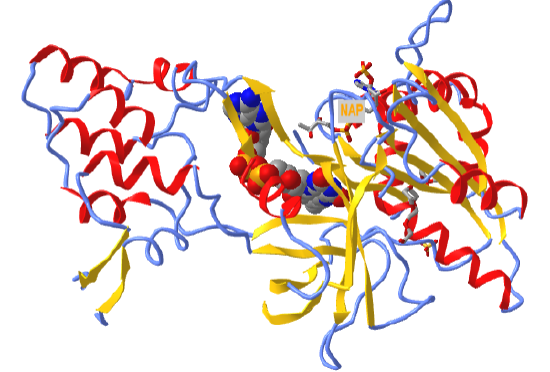
 Figure \(\PageIndex{5}\): FAD binding domain of cytochrome P450 BM3 in complex with NADP+ (4DQL). (Copyright; author via source).
Figure \(\PageIndex{5}\): FAD binding domain of cytochrome P450 BM3 in complex with NADP+ (4DQL). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?hoT1WDCUv1wZFMyRA
FAD is shown in spacefill. NADP+, which reoxidizes the reduced FADH2 back to FAD, is shown in sticks and labeled NAP.
Nicotinamide Adenine Dinucleotide (FAD) reactions
NAD+ and a phosphorylated form, NADP+, are one of nature's most widely used oxidizing agents and are used as dissociable substrates/cofactors for many different types of enzyme-catalyzed oxidation reactions. Since it binds (as a substrate) and dissociates (as a product) after each catalytic cycle, the free enzyme is continually active. The biological synthesis of NAD+ requires the vitamin nicotinic acid, also called niacin (nicotinic acid), an absence of which causes pellagra.
Oxidation of an alcohol to an aldehyde: The oxidation of ethanol to acetaldehyde by NAD+, catalyzed by the enzyme alcohol dehydrogenase, is shown in Figure \(\PageIndex{6}\).

The product acetaldehyde contributes to hangovers after ethanol consumption. Note that this reaction is a hydride transfer, which would not be expected to occur in the aqueous environment of a cell, given the extreme reactivity and basicity of a :H- hydride ion. This transfer happens in the active site of the enzyme, which is anhydrous after binding substrates.
Oxidative decarboxylation of an alcohol: A two-step mechanism for this reaction is shown in Figure \(\PageIndex{7}\)

After the first step, an electron sink (the oxygen of the carbonyl) is present at the β-carbon, facilitating the decarboxylation step.
Oxidative deamination of an amine: A two-step reaction, a hydride transfer to form a Schiff base, followed by hydrolysis of the Schiff base, is shown in Figure \(\PageIndex{8}\).

We will discuss Schiff base chemistry in more detail below.
Pyridoxal Phosphate Enzymes
Pyridoxal phosphate (PLP) is a derivative of vitamin B6 or pyridoxal. Deficiencies cause convulsions, chronic anemia, and neuropathy. It assists in many reactions (catalyzed by PLP-dependent enzymes). The PLP is bound covalently to lysine residues in a Schiff base linkage (aldimine). This form reacts with many free amino acids (as substrates) to replace the Schiff base to Lys of the enzyme with a Schiff base to the amino acid substrate. First, we will review of Schiff base (an imine) formation by the reaction of an aldehyde or ketone with an amine as shown in Figure \(\PageIndex{9}\).

The reaction is essentially a nucleophilic attack of a carbonyl carbon of an aldehyde or ketone by an amine, followed by a dehydration step. Note that the net effect is to replace one electron sink, a carbonyl (C=O), with an imine (C=NH ↔ C=NH2+), with pKa around 7.0. Hence at neutral pH, 50% of the imine is protonated to form the iminium cation, a much better electron sink than the starting carbonyl!
The structure of pyridoxal phosphate, which contains a reactive aldehyde, is converted to an imine by reaction with the ε-amino side chain of a lysine in the active site of a PLP-dependent enzyme, is shown in Figure \(\PageIndex{10}\).

The figure also shows the replacement of the enzymes' lysine ε-NH2-PLP bond to that of a free amino as an incoming substrate, a process which should proceed with a ΔG0 of approximately 0. This occurs in PLP-dependent enzymes with free amino acids as substrates (we will discuss several examples below).
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of the E. Coli Aspartate aminotransferase, W140H mutant, maleate complex (1ARI).
..png?revision=1&size=bestfit&width=394&height=412)
Figure \(\PageIndex{11}\): Aspartate aminotransferase, W140H mutant, maleate complex (1ARI). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...ZQixXhCwX5EEQ9
Note that the PLP is in Schiff base linkage with the ε-NH2 group of a lysine in the enzyme's active site.
From a chemistry perspective, PLP is an ideal molecule to facilitate electron flow in biochemical reactions. William Jencks noted this in his classic text, Catalysis in Chemistry, in which he wrote this elegant description of its properties:
"It has been said that God created an organism especially adapted to help the biologist find an answer to every question about the physiology of living systems; if this is so, it must be concluded that pyridoxal phosphate was created to provide satisfaction and enlightenment to those enzymologists and chemists who enjoy pushing electrons, for no other coenzyme is involved in such a wide variety of reactions, in both enzyme and model systems, which can be reasonably interpreted in terms of the chemical properties of the coenzyme. Most of these reactions are made possible by a common structural feature. That is, electron withdrawal toward the cationic nitrogen atom of the imine and into the electron sink of the pyridoxal ring from the alpha carbon atom of the attached amino acid activates all three of the substituents of this carbon for reactions which require electron withdrawal from this atom."
We'll present three examples of the reaction of an amino acid with a PLP-dependent enzyme. In each case, a different bond to the α-carbon of the amino acid substrate is broken.
α-decarboxylation of an amino acid: Figure \(\PageIndex{12}\) shows a plausible reaction mechanism.

β-elimination from serine: The enzyme serine dehydratase catalyzes the reaction shown in Figure \(\PageIndex{13}\).

Racemization of amino acids: Amino acid racemases use PLP as a cofactor using a mechanism shown in Figure \(\PageIndex{14}\).

Why do racemases exist since the biological world consists of only L-amino acids? There are two possible reasons. Some D-amino acids are found, such as in bacterial cell walls. In addition, amino acids spontaneously racemize on their own, albeit at a slow rate. Racemases that have oxygen atoms in the beta-carbon racemize at a much higher rate since they can stabilize the carbanion intermediate formed when the alpha proton is removed in the process of racemization. The concentration of D-Asp and D-Asn can be used in dating biological material as well.
Transamination reactions: PLP enzymes also catalyze the transamination reaction, which is shown in Figure \(\PageIndex{15}\)
Amino Acid 1 + α-keto acid 1 ↔ α-keto acid 2 + Amino Acid 2 For example: Figure \(\PageIndex{x}\)

First, Asp, bound to PLP through a Schiff base link, loses the α-H and forms a ketimine through a tautomerization reaction, which ultimately hydrolyzes to form the released oxaloacetate and pyridoxamine. The pyridoxamine reacts with α-ketoglutarate in the reverse of the first three reactions to form Glu.
We will explore other cofactors in future chapters.