
4.6: Intrinsically Disordered Proteins
- Page ID
- 62323


Intrinsically Disordered Proteins (IDPs) and Metamorphic Proteins
Many examples of proteins that are partially or completely disordered but still retain biological function have been found. At first glance this might appear to be unexpected, since how could such a protein bind its natural ligand with specificity and selectivity to express its function? Of course, one could postulate ligand binding would induce conformational changes necessary for function (such as catalysis) in an extreme example of an induced fit of a ligand compared to a "lock-and-key" fit. Decades ago, Linus Pauling predicted that antibodies, proteins that recognize foreign molecules (antigens), would bind loosely to the antigen, followed by a conformational change to form a more complementary and tighter fit. This was the easiest way to allow for a finite number of possible protein antibodies to bind a seemingly endless number of possible foreign molecules. This is indeed one method in which antibodies can recognize foreign antigens. Antibodies that bind to antigens with high affinity and hence high specificity are more likely to bind through a lock and key fit. (Pauling, however, didn't know that the genes that encode the proteins chains in antibodies are differentially spliced and subjected to enhanced mutational rates which allow the generation of incredible antibody diversity from a limited set of genes.)
Intrinsically Disordered Proteins (IDPs)
It's been estimated that over half of all native proteins have regions (greater than 30 amino acids) that are disordered, and upwards of 20% of proteins are completely disordered. Regions of disorder are enriched in polar and charged side chains which follow since these might be expected to assume many available conformations in aqueous solutions compared to sequences enriched in hydrophobic side chains, which would probably collapse into a compact core stabilized by the hydrophobic effect. Mutations in the disordered regions tend to preserve the disordered region, suggesting that the disordered region is advantageous for "future" function. In addition, mutations that cause a noncoding sequence to produce a coding one invariably produce disordered protein sequences. Disordered proteins tend to have regulatory properties and bind multiple ligands, in comparison to ordered ones, which are involved in highly specific ligand binding necessary for catalysis and transport. The intracellular concentration of disordered proteins has also been shown to be lower than ordered proteins, possibly to prevent occurrences of inappropriate binding interactions mediated through hydrophobic interactions, for example. Processes to accomplish this include more rapid mRNA and protein degradation and slower translation of mRNA for disordered proteins. For a similar reason, misfolded proteins are targeted for degradation as well. Figure \(\PageIndex{1}\) shows characteristics of intrinsically disordered proteins.
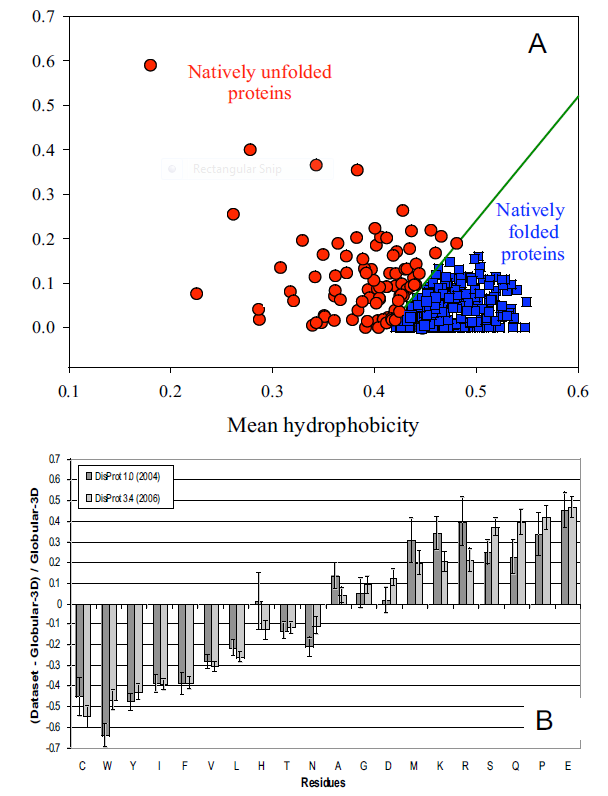
Panel A shows the mean net charge vs the mean hydrophobicity for 275 folded and 91 natively unfolded proteins. Panel B shows the relative amino acid composition of globular (ordered) proteins compared to regions of disorder greater than 10 amino acids in disordered proteins. The two different grey bars were obtained with two different versions of the software used to analyze the proteins. Again the graph shows the enrichment of hydrophilic amino acids in disordered proteins.
Many experimental methods can be used to detect disordered regions in proteins. Such regions are not resolved well in X-Ray crystal structures (have high B factors). NMR solution structures would show multiple, and differing conformations. CD spectroscopy likewise would show ill defined secondary structure. In addition solution measurements of size (light scattering, centrifugation) would show larger size distributions for a given protein.
What types of proteins contain disorder? The above experimental and new computational methods have been developed to classify proteins as to their degree of disorder. There appear to be more IDPs in eukaryotes than in archea and prokaryotes. Many IDPS are involved in cell signaling processes (when external molecules signal cells to respond by proliferating, differentiating, dying, etc). Most appear to reside in the nucleus. The largest percentage of known IDPs bind to other proteins and also to DNA. These results suggest that IEPs are essential to protein function and probably confer significant advantages to eukaryotic cells as multiple functions can be elicited from the interaction of a single IEP (derived from a single gene) with different protein binding partners. This would greatly extend the effective genome size in humans, for example, from around 20,000 protein-encoding genes with specified functions, to many more. This doesn't even take into account the increase in functionalities derived from post-translational chemical modifications.
Protein structure is fluid and complex and our simple notions and words to denote proteins as either native or denatured are misguided and constrain our ideas about how protein structure elicits biological function. For example, what does the word "native" mean, if proteins exist in multiple states in vivo and in vitro simultaneously? Dunker et al (2001) have coined the concept "Protein Trinity" to move past the notion that a single protein folds to a single state which elicits a single function. Rather each of the states in the "trinity", the ordered, collapsed (or molten globule) and extended (random coil) coexist in the cell, as shown in Figure \(\PageIndex{2}\).: Characteristics of Intrinsically Disordered Proteins. Hence all can be considered "native" and all contribute to the function of the cell. A single IDP could bind to many different protein partners, each producing different final structures and functions. IDPs would also be more accessible and hence susceptible to proteolysis, which would lead to a simple mechanism to control their concentrations, an important way to regulate their biological activity. Their propensity to post-translational chemical modification would likewise lead to new types of biological regulation.
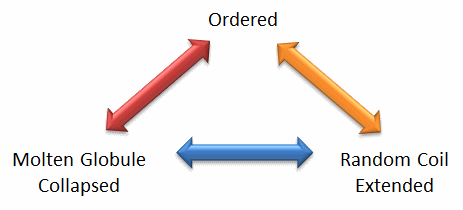
These ideas have profound ramifications for our understanding of the expression of cellular phenotype. In addition, a whole new world of drug targets is available by finding drugs that modulate the transitions between ordered, collapsed and extended protein states. Likewise, side effects of drugs might be understood by investigating their effects on these transitions in IDPs that were not initially targeted for anal. Several web database, including PONDR - Predictor of Naturally Occurring Disorder and Database of Protein Disorder are available.
IDPs cover a spectrum of states from fully unstructured to partially structured and include random coils, (pre-)molten globules, and large multi-domain proteins connected by flexible linkers. They constitute one of the main types of protein (alongside globular, fibrous and membrane proteins). Figure \(\PageIndex{3}\) shows the conformational flexibility in SUMO-1 protein (PDB:1a5r), which is a composite of 10 NMR structures. The central part shows relatively ordered structure. Conversely, the N- and C-terminal regions (left and right, respectively) show ‘intrinsic disorder’.

Figure \(\PageIndex{3}\): Conformational flexibility in SUMO-1 protein (1a5r) showing intrinsically disordered regions
It's interesting to explore the history of our understanding of IDPs. In the 1930s -1950s, the first protein structures were solved by protein crystallography. These early structures suggested that a fixed three-dimensional structure might be generally required to mediate biological functions of proteins. When stating that proteins have just one uniquely defined configuration, Mirsky and Pauling did not recognize that Fisher's work would have supported their thesis with his 'Lock and Key' model (1894). These publications solidified the central dogma of molecular biology in that the sequence determines the structure which, in turn, determines the function of proteins. In 1950, Karush wrote about 'Configurational Adaptability' contradicting all the assumptions and research in the 19th century. He was convinced that proteins have more than one configuration at the same energy level and can choose one when binding to other substrates. In the 1960s, Levinthal's paradox suggested that the systematic conformational search of a long polypeptide is unlikely to yield a single folded protein structure on biologically relevant timescales (i.e. seconds to minutes). Curiously, for many (small) proteins or protein domains, relatively rapid and efficient refolding can be observed in vitro. As stated in Anfinsen's Dogma from 1973, the fixed 3D structure of these proteins is uniquely encoded in its primary structure (the amino acid sequence), is kinetically accessible and stable under a range of (near) physiological conditions, and can therefore be considered as the native state of such "ordered" proteins.
During the subsequent decades, however, many large protein regions could not be assigned in x-ray datasets, indicating that they occupy multiple positions, which average out in electron density maps. The lack of fixed, unique positions relative to the crystal lattice suggested that these regions were "disordered". Nuclear magnetic resonance spectroscopy of proteins also demonstrated the presence of large flexible linkers and termini in many solved structural ensembles. It is now generally accepted that proteins exist as an ensemble of similar structures with some regions more constrained than others.
Some people differentiate a particular type of IDP called Intrinsically Unstructured Proteins (IUPs), which occupy the extreme end of this spectrum of flexibility, whereas IDPs also include proteins of considerable local structure tendency or flexible multidomain assemblies. These highly dynamic disordered regions of proteins have subsequently been linked to functionally important phenomena such as allosteric regulation and enzyme catalysis.
Many disordered proteins have their binding affinity with their receptors regulated by post-translational modification. Hence it has been proposed that the flexibility of disordered proteins facilitates the conformational requirements for binding their modifying enzymes as well as their receptors. Intrinsic disorder is particularly found in proteins implicated in cell signaling, transcription and chromatin remodeling functions. Here are some types or characteristics of IDPs.
Flexible linkers
Disordered regions are often found as flexible linkers or loops connecting domains. Linker sequences vary greatly in length but are typically rich in polar uncharged amino acids. Flexible linkers allow the connecting domains to freely twist and rotate to recruit their binding partners via protein domain dynamics. They also allow their binding partners to induce larger-scale conformational changes by long-range allostery.
Linear motifs
Linear motifs are short disordered segments of proteins that mediate functional interactions with other proteins or other biomolecules (RNA, DNA, sugars etc.). Many roles of linear motifs are associated with cell regulation, for instance in control of cell shape, subcellular localization of individual proteins and regulated protein turnover. Often, post-translational modifications such as phosphorylation tune the affinity (not rarely by several orders of magnitude) of individual linear motifs for specific interactions. Unlike globular proteins, IDPs do not have premade active pockets. Nevertheless, in 80% of IDPs (~3 dozen) subjected to detailed structural characterization by NMR, there are linear motifs termed PreSMos (pre-structured motifs) that are transient secondary structural elements primed for target recognition. In several cases, it has been demonstrated that these transient structures become full and stable secondary structures, e.g., helices, upon target binding. Hence, PreSMos are the putative active sites in IDPs.
Coupled folding and binding
Many unstructured proteins undergo transitions to more ordered states upon binding to their targets. The coupled folding and binding may be local, involving only a few interacting residues, or it might involve an entire protein domain. It was recently shown that the coupled folding and binding allow the burial of a large surface area that would be possible only for fully structured proteins if they were much larger. Moreover, certain disordered regions might serve as "molecular switches" in regulating certain biological functions by switching to ordered conformations upon binding small molecules, nucleic acids or ions.
Disorder in the bound state (fuzzy complexes)
Intrinsically disordered proteins can retain their conformational freedom even when they bind specifically to other proteins. The structural disorder in the bound state can be static or dynamic. In fuzzy complexes structural multiplicity is required for function and the manipulation of the bound disordered region changes activity. The conformational ensemble of the complex is modulated via post-translational modifications or protein interactions. The specificity of DNA binding proteins often depends on the length of fuzzy regions, which is varied by alternative splicing. Intrinsically disordered proteins adapt many different structures in vivo according to the cell's conditions, creating a structural or conformational ensemble.
Therefore, their structures are strongly function-related. However, only few proteins are fully disordered in their native state. Disorder is mostly found in intrinsically disordered regions (IDRs) within an otherwise well-structured protein. The term intrinsically disordered protein (IDP) therefore includes proteins that contain IDRs as well as fully disordered proteins.
The existence and kind of protein disorder is encoded in its amino acid sequence. As described above, IDPs are characterized by a low content of bulky hydrophobic amino acids and a high proportion of polar and charged amino acids, usually referred to as low hydrophobicity. This property leads to good interactions with water. Furthermore, high net charges promote disorder because of electrostatic repulsion resulting from equally charged residues. Thus disordered sequences cannot sufficiently bury a hydrophobic core to fold into stable globular proteins. In some cases, hydrophobic clusters in disordered sequences provide clues for identifying the regions that undergo coupled folding and binding (refer to biological roles).
Many disordered proteins reveal regions without any regular secondary structure These regions can be termed as flexible, compared to structured loops. While the latter are rigid and contain only one set of Ramachandran angles, IDPs involve multiple sets of angles. The term flexibility is also used for well-structured proteins, but describes a different phenomenon in the context of disordered proteins. Flexibility in structured proteins is bound to an equilibrium state, while it is not so in IDPs. Many disordered proteins also reveal low complexity sequences, i.e. sequences with over-representation of a few residues. While low complexity sequences are a strong indication of disorder, the reverse is not necessarily true, that is, not all disordered proteins have low complexity sequences. Disordered proteins have a low content of predicted secondary structure.
Silent Single nucleotide polymorphisms (SNPs)
For some amino acids, multiple triplet nucleotide sequences (codons) in the coding regions of a gene for a protein lead to the incorporation of the same amino acid in the protein sequence. Hence two proteins identical in amino acid sequence might have slightly different nucleotide sequences in the gene that encodes them. Such single nucleotide polymorphisms (SNPs) in coding regions were thought to have no effect on the tertiary structure and biological function of a protein if the single nucleotide variation did not lead to the insertion of a different amino acid into the growing peptide chain (i.e the codons were synonymous and the mutations presumably silent with no effect). Recently single nucleotide polymorphisms (SNPs) in the gene for the product of the MDR1 (multidrug resistance 1) gene, P-glycoprotein, was shown to result in a protein with different substrate specificity and inhibitor interactions, and hence a different 3D structure. One possible explanation for this observation is a difference in the rate of translation of the mRNA for this membrane protein. Different rates might lead to different intra- and intermolecular associations, which could lead to different final 3D structures as the protein cotranslationally folds and inserts into the membrane. This would especially be true if two possible structures were close enough in free energy but separated by a significant activation energy barrier, precluding simple conformational rearrangement of one conformation to another.
It has been shown in yeast that synonymous mutations (those that don't change the amino acid on mutation of the DNA encoding the particular amino acid) generally have the same effect on the "health" of yeast as do non-synonymous mutations (those that change the amino acid). This rather startling result upends much dogma. Some possible expected effects of synonymous mutation include alteration in gene expression of the mutated gene and possible effects on the stability of the transcribed RNA from the mutated RNA. mRNA levels are lowered from both types of mutations as well as fitness levels of the yeast, as defined by speed of growth.
Metamorphic Proteins
In addition to prion proteins, it appears that many proteins can adopt more than one conformation under the same set of conditions. In contrast to prion proteins, however, in which the formation of the beta-structure variant is irreversible since the conformational change is associated with aggregation, many proteins can change conformations reversibly. Often, these changes do not appear to be associated only with binding interactions that trigger the change. Murzin has described proteins that change conformations on change of the pH (viral glycoproteins), redox state (chloride channel), disulfide isomerization (lysozyme), and bound ligand (RNA polymerase as it initiates and then elongates the growing RNA polymer). He cites two proteins that appear to change state without external signals. These include Mad2, in which the two conformers share an extensive similarity, and Ltn10 (lymphotactin), in which they don't. One form of lymphotactin (Ltn 10) binds to similar lymphokine receptors, while the other (Ltn 40) binds to heparin. Folding kinetics may play a part in these examples as well, as proteins capable of folding to two conformers independently and quickly might prevent misfolding and aggregation that might occur if they had to completely unfold first before a conformational transition. Both Mad2 and Ltn10 alter conformation through transient formations of dimers, which facilitate conformational changes without widespread unfolding. Mutations in Ltn10 can cause the protein to adopt the Ltn40 conformation, Hence primordial "metamorphic" proteins could, by simple mutation, produce new protein functionalities.
Metamorphic proteins, which display large structural changes, usually involving large changes in hydrogen bonding and hence secondary structure, are different from simpler allosteric proteins chose conformation changes are smaller. Few metamorphic proteins have been found but some speculate they could account for as much as 5% of proteins. A wonderful example of such a protein is the human chemokine XCL1 (lymphotactin) protein, an immune regulatory protein. It undergoes a huge transition from a form that has a typical chemokine fold to a dimer that has an extensive beta structure. Ltn lacks one of the two disulfide bonds found in all other chemokines, which allows greater conformation flexibility.
Figure \(\PageIndex{3}\) shows interactive iCn3D model of the solution structures of monomeric and XCL1 (lymphotactin, PDB 2HDM)
| Monomeric XCL1 (lymphotactin PDB 2HDM) | Dimeric XCL1 (lymphotactin PDB 2JP1) |
|
(Copyright; author via source). |
(Copyright; author via source). |
.png?revision=1)
.png?revision=1)
References
f17f4df-605c-4388-88c2-25b0f000b0ed@2.
File:Chirality with hands.jpg. (2017, September 16). Wikimedia Commons, the free media repository. Retrieved 17:34, July 10, 2019 from commons.wikimedia.org/w/index.php?title=File:Chirality_with_hands.jpg&oldid=258750003.
Wikipedia contributors. (2019, July 6). Zwitterion. In Wikipedia, The Free Encyclopedia. Retrieved 21:48, July 10, 2019, from en.Wikipedia.org/w/index.php?title=Zwitterion&oldid=905089721
Wikipedia contributors. (2019, July 8). Absolute configuration. In Wikipedia, The Free Encyclopedia. Retrieved 15:28, July 14, 2019, from en.Wikipedia.org/w/index.php?title=Absolute_configuration&oldid=905412423
Structural Biochemistry/Enzyme/Active Site. (2019, July 1). Wikibooks, The Free Textbook Project. Retrieved 16:55, July 16, 2019 from en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Enzyme/Active_Site&oldid=3555410.
Structural Biochemistry/Proteins. (2019, March 24). Wikibooks, The Free Textbook Project. Retrieved 19:16, July 18, 2019 from en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Proteins&oldid=3529061.
Fujiwara, K., Toda, H., and Ikeguchi, M. (2012) Dependence of a α-helical and β-sheet amino acid propensities on teh overall protein fold type. BMC Structural Biology 12:18. Available at: https://bmcstructbiol.biomedcentral.com/track/pdf/10.1186/1472-6807-12-18
Wikipedia contributors. (2019, July 16). Keratin. In Wikipedia, The Free Encyclopedia. Retrieved 17:50, July 19, 2019, from en.Wikipedia.org/w/index.php?title=Keratin&oldid=906578340
Wikipedia contributors. (2019, July 13). Alpha-keratin. In Wikipedia, The Free Encyclopedia. Retrieved 18:17, July 19, 2019, from en.Wikipedia.org/w/index.php?title=Alpha-keratin&oldid=906117410
Open Learning Initiative. (2019) Integumentary Levels of Organization. Carnegie Mellon University. In Anatomy & Physiology. Available at: https://oli.cmu.edu/jcourse/webui/syllabus/module.do?context=4348901580020ca6010f804da8baf7ba.
Wikipedia contributors. (2019, July 16). Collagen. In Wikipedia, The Free Encyclopedia. Retrieved 03:42, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Collagen&oldid=906509954
Wikipedia contributors. (2019, July 2). Rossmann fold. In Wikipedia, The Free Encyclopedia. Retrieved 16:01, July 20, 2019, from https://en.Wikipedia.org/w/index.php?title=Rossmann_fold&oldid=904468788
Wikipedia contributors. (2019, May 30). TIM barrel. In Wikipedia, The Free Encyclopedia. Retrieved 16:46, July 20, 2019, from en.Wikipedia.org/w/index.php?title=TIM_barrel&oldid=899459569
Wikipedia contributors. (2019, July 16). Protein folding. In Wikipedia, The Free Encyclopedia. Retrieved 18:30, July 20, 2019, from https://en.Wikipedia.org/w/index.php?title=Protein_folding&oldid=906604145
Wikipedia contributors. (2019, June 11). Globular protein. In Wikipedia, The Free Encyclopedia. Retrieved 18:49, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Globular_protein&oldid=901360467
Wikipedia contributors. (2019, July 11). Intrinsically disordered proteins. In Wikipedia, The Free Encyclopedia. Retrieved 19:52, July 20, 2019, from en.Wikipedia.org/w/index.php?title=Intrinsically_disordered_proteins&oldid=905782287
- Comprehensive Database for Protein Analysis - Biozon
- SCOP: Structural Characterization of Proteins - Database showing folds, superfamiles, families, and domains