
1.3: Physical-Chemical Foundations
- Page ID
- 102242


\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)(Learning goals written by Claude, Sonnet 4.6, Anthropic)
Reaction Extent, Equilibrium, and the Equilibrium Constant
- Distinguish among irreversible, partially reversible, and reversible reactions by interpreting concentration-vs-time profiles, and relate the magnitude and direction of Keq to the intrinsic relative stability of reactants and products.
- Apply Le Châtelier's Principle to predict how perturbations in concentration, temperature, or product removal shift a reaction at equilibrium, and explain why cells exploit these principles to drive otherwise unfavorable reactions forward.
Gibbs Free Energy and Reaction Spontaneity
- Write and interpret the equation ΔG = ΔG° + RT ln Q, identifying the separate contributions of intrinsic molecular stability (ΔG°) and concentration (RT ln Q) to the overall driving force for a reaction.
- State the relationship between ΔG° and Keq at equilibrium, explain why ΔG° is concentration-independent, and justify the use of ΔG°' (at pH 7) as the relevant standard for biochemical reactions.
- Use the sign of ΔG to predict reaction direction, recognizing that ΔG = 0 defines equilibrium and that ΔG evolves continuously as concentrations change until equilibrium is reached.
Enthalpy, Entropy, and the Thermodynamic Basis of Spontaneity
- Define enthalpy and entropy in thermodynamically precise terms, distinguishing exothermic from endothermic reactions and explaining why heat release alone does not determine spontaneity.
- Explain entropy as a measure of the dispersal of energy and matter across microstates (via Boltzmann's equation S = k ln W), and avoid the misleading macroscopic "disorder" framing by grounding the concept in positional and thermal entropy contributions.
- Derive the Gibbs free energy function G = H − TS from the requirement that total entropy of the universe must increase for any spontaneous process, and use ΔG = ΔH − TΔS to evaluate how enthalpy and entropy changes combine to determine spontaneity under conditions of constant temperature and pressure.
The types and numbers of chemical reactions in biological cells are staggering. Compared to physical and chemical reactions in a controlled and closed environment, biological reactions occur in open systems with input and output of energy and chemical "feedstocks." Yet, they are governed by the same physical principles that control all reactions. We can gain insight into biological reactions and how they are controlled by considering the principles you have used in many preceding classes: energy changes, equilibria, and thermodynamics. Let's review them!
Reactions and Energy Changes
Why do reactions vary in extent from completely irreversible in the forward reaction to reversible reactions favoring the reactants? It may be helpful to understand a simple physical reaction before attempting more complex chemical reactions. Let's start with a simple ball on a hill. Does a ball at the top of a hill roll downhill spontaneously, or does the opposite happen? No one has ever seen a ball roll spontaneously uphill unless a lot of energy was added to the ball. This physical reaction appears irreversible and is driven to the lower potential energy at the bottom of the hill. The potential energy gap is related to the "extent" and spontaneity of this reaction. As we have undoubtedly observed, natural processes tend to go to a lower energy state. By analogy, we will consider the driving force for a chemical reaction to be the free energy difference, ΔG, between reactants and products. ΔG determines the extent and spontaneity of the reaction.
Reversible/Irreversible Reactions, Extent of Reactions, Equilibria
Consider a hypothetical reversible reaction in which you start with some reactants, \(\ce{A}\) and \(\ce{B}\), each at a 1 M concentration (1 mol of each/L solution). but no products, \(\ce{P}\) and \(\ce{Q}\). For ease, assume that the total volume of solution is 1 L, so that we start with 1 mol each of \(\ce{A}\) and \(\ce{B}\). At time \(t=0\), the concentration of products is 0. The reaction can be written as:
\[\ce{A + B <=> P + Q}. \nonumber \]
As time progresses, the amounts or concentrations of \(\ce{A}\) and \(\ce{B}\) decrease as the amounts or concentrations of products \(\ce{P}\) and \(\ce{Q}\) increase. At some point, no further changes occur in the amounts or concentrations of the remaining reactants or products. At this point, the reaction is in equilibrium, a term often used in our common vocabulary to denote a system undergoing no net change.
Most of the reactions we will study occur in solution, so we will deal with concentrations in mol/L or mmol/mL (M). Let's consider how the concentrations of reactants and products change over time. Depending on the extent to which a reaction is reversible, four different scenarios can be imagined:
Scenario 1: Irreversible reaction in which the reverse reaction occurs to a negligible extent.
In this reaction, the reverse reaction occurs to such a small extent that it can be neglected. The only reaction that occurs is the conversion of reactants to products. Hence, all the reactants are converted into the product. At equilibrium, [A] = 0. Since 1 mol of A reacted, it must form 1 mol of P and 1 mol of Q - i.e., the concentration of products at equilibrium is 1 M. At an earlier time of the reaction (let's pick a time when [A] = 0.8 M), only part of the reactants have reacted (in this case, 0.2 M), producing an equal amount of products, P and Q. Graphs of [A] and [P] as a function of time are shown below. [A] decreases nonlinearly to 0 M while [P] increases reciprocally to 1 M concentration. This is illustrated in the graph below.
![Graph showing the concentration of reactants [A] in blue decreasing over time, while the concentration of product [P] in red increases.](https://bio.libretexts.org/@api/deki/files/101209/clipboard_efabf117bf5204a5bb907499652514319.png?revision=1)
Examples of irreversible reactions include those of strong acids (such as nitric, sulfuric, and hydrochloric) with bases (OH- and water), as well as the much more complex combustion reactions, such as the burning of sugars (like those found in trees) and hydrocarbons (like octane) to form CO2 and H2O.
Scenario 2: Reversible reaction in which the forward reaction is favored.
Again, [A] decreases and [P] increases, but in this case, some [A] remains because the reaction is reversible. As [A ]and [B] decrease, [P] and [Q] increase, which increases the chance that they will collide and form the product. Since P and Q can react to form reactants, the [A] at equilibrium is not zero, as is shown below.
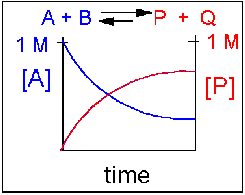
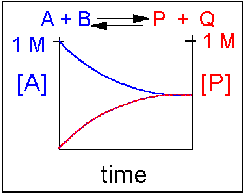
Scenario 3: Reversible Reaction in which forward and reverse reactions are equally favored.
Again, [A] decreases and [P] increases, but in this case, some [A] remains because the reaction is reversible. As [A] and [B] decrease, [P] and [Q] increase, thereby increasing the likelihood that they will collide and form the product. Since P and Q can react to form reactants, the [A] at equilibrium is not zero, as is shown below. Because the reactants and products are equally favored, their concentrations will be equal at equilibrium.
Scenario 4: Reversible Reaction in which the reverse reaction is favored.
Again, [A] decreases, and [P] increases, but in this case, some A remains since the reaction is reversible. As [A] and [B] decrease, [P] and [Q] increase, thereby increasing the likelihood that they will collide and form the product. Since P and Q can react to form reactants, the [A] at equilibrium is not zero, as shown below. Because the reaction favors the reactants, their concentration will be higher at equilibrium than that of the products.
![Graph showing the concentrations of reactants [A] (blue) and products [P] (red) over time in a chemical equilibrium reaction.](https://bio.libretexts.org/@api/deki/files/50055/revrx4.gif?revision=1)
An example of this kind of reaction, which favors the reactants, is the reaction of acetic acid (a weak acid) with water.
\[\ce{CH3CO2H(aq) + H2O(l) <=> H3O^{+}(aq) + CH3CO2^{-}(aq)} \nonumber \]
Equilibrium Constants
Without much experience in chemistry, it is challenging to examine the structures of the reactants and products and determine whether the reaction is irreversible or reversible, favoring either the reactants or the products (except for obvious irreversible reactions described above). However, this data can be found in tables of equilibrium constants. The equilibrium constant, as its name implies, is constant, independent of the concentration of the reactants and products. A \(K_{eq} > 1\) implies that the products are favored. A \(K_{eq} < 1\) implies that reactants are favored. Reactants and products are equally favored when \(K_{eq} = 1\). For the more general reaction,
\[\ce{aA + bB <=> pP + qQ} \nonumber \]
where \(a\), \(b\), \(p\), and \(q\) are the stoichiometric coefficients,
\begin{equation}
\mathrm{K}_{\mathrm{eq}}=\frac{[\mathrm{P}]_{\mathrm{eq}}^{\mathrm{p}}[\mathrm{Q}]_{\mathrm{eq}}^{\mathrm{q}}}{[\mathrm{A}]_{\mathrm{eq}}^{\mathrm{a}}[\mathrm{B}]_{\mathrm{eq}}^{\mathrm{b}}}
\end{equation}
where all the concentrations are at their equilibrium values. For a simple reaction where \(a\), \(b\), \(p\), and \(q\) are all 1, then \(K_{eq} = ([P] [Q])/([A] [B]) \).
(Note: Equilibrium constants are truly constant only at a given temperature, pressure, and solvent condition. Likewise, they depend on concentration to the extent that their activities change with concentration.)
For an irreversible reaction, such as the reaction of 0.1 M HCl (aq) in water, [HCl]eq = 0, you cannot easily measure a Keq. However, if we assume the reaction goes in reverse to an almost imperceptible degree, [HCl]eq might equal 10-10 M. Hence Keq >> 1.
In summary, the extent of different reactions can range from completely irreversible (favoring only the products) to reversible (favoring the reactants).
Our next goal is to understand what controls the extent of a reaction. That is, of course, the change in the Gibbs free energy. Two different pairs of factors influence the ΔG. One pair is the reactants' concentration and their inherent reactivity relative to the products (as reflected in the Keq). The other pair is enthalpy/entropy changes. We will now consider the first pair.
Contributions of Molecule Stability (Keq) and Concentration to ΔG
Consider the reactions of hydrochloric acid and acetic acid with water.
\[\begin{align*} \ce{HCl (aq) + H2O (l)} & \ce{-> H3O^{+}(aq) + Cl^{-} (aq)} \\[4pt] \ce{CH3CO2H (aq) + H2O (l) } & \ce{-> H3O^{+} (aq) + CH3CO2^{}- (aq)} \end{align*} \nonumber \]
Assume that at t = 0, each acid is placed into water at a concentration of 0.1 M. When equilibrium is reached, essentially no HCl remains in the solution. In contrast, 99% of the acetic acid remains. Why are they so different? We rationalized that HCl (aq) is a much stronger acid than H3O+(aq), which itself is a much stronger acid than CH3CO2H (aq). Why? Something about the structure of these acids (and the bases) makes HCl much more intrinsically unstable, much higher in energy, and hence much more reactive than the acid it forms, H3O+(aq). Likewise, H3O+(aq) is much more intrinsically unstable, much higher in energy, and hence more reactive than CH3CO2H (aq). This has nothing to do with concentration, since the initial concentration of both HCl (aq) and CH3CO2H (aq) was identical. This observation is reflected in the Keq for these acids (>>1 for HCl and <<1 for acetic acid). This difference in the intrinsic stability of reactants relative to products (independent of concentration) is one factor contributing to ΔG.
The other factor is concentration. A 0.25 M (0.25 mol/L or 0.25 mmol/ml) solution of acetic acid does not conduct electricity, implying that very few ions of H3O+(aq) + CH3CO2- (aq) exist in the solution. However, a dim light becomes evident if more concentrated acetic acid is added. Adding more reactants appeared to drive the reaction toward more products, even though the reverse reaction is favored based on the intrinsic stability of reactants and products alone. Before the concentrated acid was added, the system was at equilibrium. Adding concentrated acid perturbed the equilibrium, which drove the reaction to form additional products. This is an example of Le Châtelier's Principle, which states that if a reaction at equilibrium is perturbed, the reaction will shift in the direction that counteracts the perturbation. Hence, if
- more reactant is added, the reaction shifts to form more products
- more product is added, the reaction shifts to form more reactants
- products are selectively removed (by distillation, crystallization, or further reaction to produce another species), the reaction shifts to form more products.
- reactants are removed (as above), the reaction shifts to form more reactants.
- heat is added to an exothermic reaction, the reaction shifts to remove the excess heat by shifting to form more reactants. (opposite for an endothermic reaction).
Change in Free Energy G
Without doing a complicated derivation, these simple examples suggest that the total \(ΔG\) can be expressed as the sum of the two contributions, showing the effects of the intrinsic stability (\(K_{eq}\)) and concentration:
\begin{equation}
\Delta G_{\text {total }}=\Delta \mathrm{G}_{\text {stability }}+\Delta \mathrm{G}_{\text {concentration }}
\end{equation}
which becomes for the simple reaction \(\ce{A + B <=> P + Q}\) (after a rigorous derivation):
\begin{equation}
\begin{aligned}
\Delta G &=\Delta G^0+R T \ln \frac{[\mathrm{P}][\mathrm{Q}]}{[\mathrm{A}][\mathrm{B}]} \\
&=\Delta G^0+R T \ln \mathrm{Q}_{\mathrm{rx}}
\end{aligned}
\end{equation}
where ΔGo reflects the contribution from the relative intrinsic stability of reactants and products, and the second term reflects the contribution from the relative concentrations of reactants and products (which has nothing to do with stability). Qrx is the reaction quotient, which for the reaction A + B ↔ P + Q is given by:
\begin{equation}
Q_{r x}=([P][Q]) /([A][B])
\end{equation}
at any point in the reaction.
Remember that ΔG is the "driving" force for a reaction, analogous to the difference in potential energy for a ball on a hill. Go back to that analogy. If the ball starts at the top of the hill, does it roll downhill? Of course. It transitions from high to low potential energy. The reaction can be written as:
Ball top → Ball bottom
for which the change in potential energy, ΔPE = PEbottom -PEtop< 0. If the ball starts at the bottom, will it go to the top? Obviously not. For that reaction,
Ball bottom → Ball top, ΔPE > 0.
If the top of the hill were at the same height as the bottom of the hill (obviously an absurd situation), the ball would not move. It would effectively be at equilibrium, a state of no change. For this reaction, Ball top --> Ball bottom, the ΔPE = 0. As the ball starts rolling down the hill, its potential energy approaches the potential it would have at the bottom. Hence, the ΔPE changes from negative to increasingly positive until it reaches its minimum value, at which point ΔPE = 0, and movement ceases. If the ΔPE is not 0, the ball will move until the ΔPE = 0.
Likewise, for a chemical reaction that favors products, ΔG < 0. The system is not at equilibrium, and the reaction will go toward products. As the reaction proceeds, products build up, and there is less of a driving force for reactants to go to products (Le Chatelier's Principle), so the ΔG becomes more positive until the ΔG = 0, and the reaction is at equilibrium. A reaction with a ΔG > 0 is likewise not at equilibrium, so it will go in the appropriate direction until equilibrium is reached. Hence, for the reaction A + B <==> P + Q,
- if ΔG < 0, the reaction goes toward products P and Q
- if ΔG = 0, the reaction is at equilibrium, and no further change occurs in the concentration of reactants and products.
- if ΔG > 0, the reaction goes toward reactants A and B.
We cannot easily measure the actual free energy (G) of reactants or products, but we can readily determine ΔG. These points are illustrated in the interactive graph below of ΔG vs time for the hypothetical reaction A + B ↔ P + Q. The blue line represents the ΔG for the reaction (A + B ↔ P + Q) when it is driven to the right by concentration changes, while the red line shows the ΔG when the reaction is driven to the left by concentration changes.
Notice that ΔG changes continuously until the system reaches equilibrium. Initially, the equilibrium is perturbed, causing the system to deviate from equilibrium (shown in blue). The perturbation favored the products. After reaching equilibrium, the system was perturbed again to favor the reverse reaction. Notice, in this case, the ΔG for the reaction as written: A + B ↔ P + Q is positive - i.e., it is not in equilibrium. Therefore, the reaction (as written) goes backward to reactants. It is essential to recognize that the reported ΔG represents the reaction as written.
Now let's apply ΔG = ΔGo + RTln Q = ΔGo + RTln ([P][Q])/([A][B]) to two reactions we discussed above:
- HCl(aq) + H2O(l) ↔ H3O+(aq) + Cl-(aq)
- CH3CO2H(aq) + H2O(l) ↔ H3O+(aq) + CH3CO2-(aq)
At time t = 0, 0.1 mol of HCl and CH3CO2H were added to two different beakers. The forward reaction is favored at this point, but obviously to different extents. The RTln Q would be identical for both acids, since each reactant is at 0.1 M, and no products exist. However, the ΔGo is negative for HCl and positive for acetic acid since HCl is a strong acid. Hence, at t=0, ΔG for the HCl reaction is much more negative than for acetic acid. This is summarized in the table below. The direction of the arrow shows if products (-->) or reactants (<---) are favored. The size of the arrow indicates approximately the extent to which the ΔG term is favored
| Reaction at t=0 | ΔGo | RTln Q | ΔG |
| HCl(aq) + H2O(l) | ---------------> | ---------------> | -----------------------------> |
| CH3CO2H(aq) + H2O(l) | <------------- | ---------------> | -> |
Now, when equilibrium is reached, no net change occurs in the concentration of reactants and products, and ΔG = 0. In the case of HCl, there is just an infinitesimal amount of HCl left and 0.1 M of each product, so concentration favors HCl formation. However, the intrinsic relative stability of reactants and products still favors products. In the case of acetic acid, most acetic acid remains (0.099 M) with little product (0.001 M), so concentration favors products. However, the intrinsic relative stability of reactants and products still favors reactants. This is summarized in the table below.
| Reaction at equilibrium. | ΔGostab | RTln Q | ΔG |
| HCl(aq) + H2O(l) | ---------------> | <--------------- | favors neither, = 0 |
| CH3CO2H(aq) + H2O(l) | <------------- | --------------> | favors neither, = 0 |
Compare the two tables above (one at time t=0 and the other at equilibrium). Notice:
- ΔGo does not change in a given set of conditions since it has nothing to do with concentration.
- Only RTln Q changes during a reaction until equilibrium is achieved.
Meaning of ΔGo
To get a better understanding of the significance of ΔGo, let's consider the following equations under two different conditions:
\begin{equation}
\Delta G=\Delta G^0+R T \ln \frac{[\mathrm{P}][\mathrm{Q}]}{[\mathrm{A}][\mathrm{B}]}=\Delta G^0+R T \ln \mathrm{Q}_{\mathrm{rx}}
\end{equation}
Condition I: Reaction at equilibrium, ΔG = 0
The equation reduces to:
\begin{equation}
\Delta G^0=-R T \ln \frac{[\mathrm{P}]_{\mathrm{eq}}[\mathrm{Q}]_{\mathrm{eq}}}{[\mathrm{A}]_{\mathrm{eq}}[\mathrm{B}]_{\mathrm{eq}}}=-2.303 \mathrm{R} T \log \mathrm{K}_{\mathrm{eq}}
\end{equation}
This supports our idea that ΔGo is independent of concentration since Keq should also be independent of concentration.
Condition II: The concentration of all reactants and products is 1 M (standard state, assuming solution reaction)
The equation reduces to:
\begin{equation}
\begin{aligned}
\Delta G=\Delta G^0+R T \ln \frac{[1][1]}{[1][1]}=\Delta G^0+2.303 R T \log 1=\Delta G^0 \\
\Delta G &=\Delta G^o+R T \ln \left(\frac{[1][1]}{[1][1]}\right) \\
&=\Delta G^o+2.303 R T \log 1 \\
&=\Delta G^o
\end{aligned}
\end{equation}
This implies that when all reactants are at this concentration, defined as the standard state (1 M for solutes), the ΔG at that particular moment is the ΔGo for the reaction. If one of the reactants or products is H3O+, it would make little biological sense to calculate ΔGo for the reaction using the standard state of [H3O+] = 1 M or a pH of zero. Instead, it is assumed that the pH = 7, [H3O+] = 10-7 M. A new symbol is used for ΔGo under these conditions, ΔGo'.
Heat, Enthalpy, and Entropy
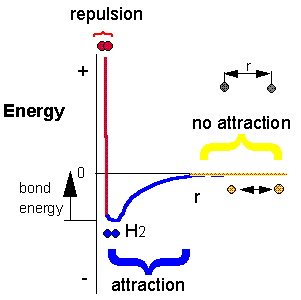
Consider the association reaction of hydrogen atoms into molecular hydrogen
\[\ce{H + H -> H2}. \nonumber \]
Does this reaction occur spontaneously? It does. You should remember that individual \(\ce{H}\) atoms are unstable since they don't have a completed outer shell of electrons - in this case, a duet. As they approach, they can interact to form a covalent bond, releasing energy in the process. The bonded state is lower in energy than two separated H atoms. This should be clear since energy has to be added to a molecule of \(\ce{H2}\) to break the bond. We refer to this as the bond dissociation energy.
`
Now consider a more complicated reaction, the burning of octane.
\[\ce{2C8H18(l) + 25O2(g) → 16CO2(g) + 18H2O(g)} \nonumber \]
To carry out this reaction, every C-C, C-H, and O-O bond in the reactants must be broken (each of which requires an input of energy), but energy is released when the products' C-O and H-O covalent bonds are formed. Is more energy needed to break the bonds in the reactants, or is more energy released when forming bonds in the product? The answer should be clear. The products must have a lower energy than the reactants, as large amounts of heat and light energy are released during the combustion of gasoline and other hydrocarbons.
These reactions suggest that energy must be released for a reaction to proceed at all in a given direction.
Now consider, however, the following reaction:
\[\ce{Ba(OH)2. 8H2O(s) + 2NH4SCN(s) -> 10H2O(l) + 2NH3(g) + Ba(SCN)2(aq)} \nonumber \]
When these two solids are added to a beaker and stirred, a reaction occurs, as evidenced by the formation of a liquid (water) and the smell of ammonia. Surprisingly, heat is not released into the surroundings in this reaction. Instead, heat was absorbed from the surroundings, causing the beaker to become so cold that it froze to a piece of wood (with a layer of water added to the wood) on which it was placed. This reaction appears to contradict our understanding that a reaction proceeds in a direction where heat is liberated. Reactions that liberate heat and raise the temperature of the surroundings are called exothermic reactions. Reactions that absorb heat from the surroundings and, hence, lower the surroundings' temperature are endothermic reactions. To answer the question, we need to consider entropy.
A review of thermodynamics
You may remember from General Chemistry that the change in the internal energy of a system, \(ΔE\), is given by:
\begin{equation}
\begin{aligned}
\Delta E_{s y s} &=q+w \\
&=q-P_{e x t} \Delta V
\end{aligned}
\end{equation}
where \(q\) is the heat (thermal energy) transferred to (+) or from the system (-), \(w\) is the work done on (+) or by (-) the system. This is one expression for the 1st Law of Thermodynamics
If only pressure/volume (PV) work is done (and not electrical work, for example), \(w = - P_{ext}ΔV\), where \(P_{ext}\) is the external pressure resisting a volume change in the system, \(ΔV\). Under these conditions, the heat transfer at constant \(P\), \(q_P\) is given by:
\begin{equation}
\begin{aligned}
\Delta \mathrm{E}_{\mathrm{sys}}-\mathrm{w} &=\Delta \mathrm{E}_{\mathrm{sys}}+\mathrm{P}_{\mathrm{ext}} \Delta \mathrm{V} \\
&=\mathrm{q}_{\mathrm{P}} \\
&=\Delta \mathrm{H}_{\mathrm{sys}}
\end{aligned}
\end{equation}
\(q_p\), which can easily be measured in a coffee cup calorimeter, is equal to the change in enthalpy, \(ΔH\), of the system.
In exothermic reactions, the reactants have more thermal energy than the products, and the heat released (measured in kilocalories or kilojoules) is the difference between the energy of the reactants and the products. When heat energy is used to measure the difference in energy, we refer to the energy as enthalpy (\(H\)) and the heat released as the change in enthalpy (\(ΔH\)), as illustrated below.
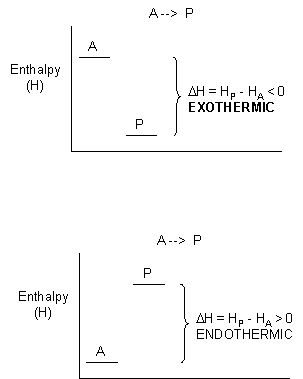
For exothermic reactions, \(ΔH < 0\). For endothermic reactions, \(ΔH > 0\).
The equation \(\Delta \mathrm{E}_{\mathrm{sys}}=\mathrm{q}+\mathrm{w}=\mathrm{q}-\mathrm{P}_{\mathrm{ext}} \Delta \mathrm{V}\) is one expression of the First Law of Thermodynamics. Another statement of energy conservation is:
\begin{equation}
\Delta \mathrm{E}_{\text {tot }}=\Delta \mathrm{E}_{\text {universe }}=\Delta \mathrm{E}_{\text {sys }}+\Delta \mathrm{E}_{\text {surrounding }}=0
\end{equation}
Something other than heat being released from the system decides whether a reaction proceeds to a significant extent. That is, the spontaneity of a reaction must depend on more than just ΔHsys. Another example of a spontaneous natural reaction is the evaporation of water (a physical, not chemical, process).
\[\ce{H2O (l) → H2O (g)} \nonumber \]
Heat is absorbed from the surroundings to break the intermolecular forces (H-bonds) among the water molecules (the system), thereby turning the liquid into a gas. If the surroundings are the skin, evaporating sweat cools the body. Why are these reactions spontaneous and essentially irreversible even though they are endothermic? Notice that in both of these endothermic reactions (the reactions of Ba(OH)2.8H2O(s) and 2NH4SCN(s) and the evaporation of water), the products are more disorganized (more disordered) than the reactants. A solid is more ordered than a liquid or a gas, and a liquid is more ordered than a gas. In nature, ordered things tend to become more disordered over time. Entropy (S), the other factor (in addition to enthalpy changes), is often considered to measure a system's disorder. The greater the entropy, the greater the disorder. For reactions that go from order (low S) to disorder (high S), the change in S, ΔS > 0. For the reaction that proceeds from low order to high order, ΔS < 0.
However, this common description of entropy is quite misleading. Macroscopic examples describing ordered/disordered states (such as the cleanliness of your room or the shuffling of a deck of cards) are inappropriate since entropy deals with microscopic states.
The driving force for spontaneous reactions is the dispersion of energy and matter. Increases in entropy for reactions involving matter occur when gases or solutes in solution are dispersed, thereby increasing positional entropy. For reactions involving energy changes, entropy increases when energy is dispersed as random, undirected thermal motion, increasing thermal entropy. In this sense, entropy, \(S\) (a measure of "spreadedness"), is a measure of the number of different ways (microstates) that particles or energy can be arranged (W), not a measure of disorder! W is an abbreviation for the German word Wahrscheinlichkeit, which means probability. It can be shown that for a solute dissolving in a solvent, Wsys = Wsolute x Wsolvent. Note that this is a multiplicative function. Entropy is a logarithmic function of W, allowing the additivity of solute and solvent W values —a feature also found in other thermodynamic state functions, such as ΔE, ΔH, and ΔS. Hence
\begin{equation}
\ln W \text { sys }=\ln W_{\text {solute }}+\ln W_{\text {solvent }}
\end{equation}
Boltzmann showed that for molecules,
\begin{equation}
S=k \ln W
\end{equation}
where \(k\) is the Boltzmann constant (1.68 x 10-23 J/K), S units: J/K
or
\begin{equation}
S=k N_A \ln W=R \ln W
\end{equation}
Boltzmann realized the connection between the macroscopic entropy of a system and the microscopic order/disorder of a system through the equation \(S = k\ln W\). Increasing S (macroscopic property) occurs with an increasing number of possible microscopic states for the atoms and molecules in a system.
The dissolution of a solute in water and the expansion of a gas into a vacuum, both of which proceed spontaneously toward an increase in matter dispersal, are examples of familiar processes characterized by ΔSsys > 0. We will see in future chapters that entropy changes in the solvent, solutes, and protein are critical determinants of protein folding.
The spontaneity of exothermic and endothermic processes will depend on the
\begin{equation}
\Delta S_{t o t}=\Delta S_{\text {surr }}+\Delta S_{\text {sys }}
\end{equation}
ΔSsys often depends on matter dispersal (positional entropy). ΔSsurr depends on energy changes in the surroundings, ΔHsurr = -ΔHsys (thermal entropy).
It is more convenient to express thermodynamic properties in terms of the system being studied rather than its surroundings. This can be readily done for ΔSsurr, which depends on ΔHsys and the temperature. First, consider the dependency on ΔHsys. Thermal energy flows into or out of the system, and since ΔHsys = - ΔHsurr,
\(ΔS_{surr}\) is proportional to -ΔHsys
- For an exothermic reaction, ΔSsurr > 0 (since ΔHsys < 0) and the reaction is favored;
- For an endothermic reaction, ΔSsurr < 0, (since ΔHsys > 0), and the reaction is disfavored;
\(ΔS_{surr}\) also depends on the temperature T of the surroundings:
\(ΔS_{surr}\) is proportional to 1/T
If the Tsurr is high, a given heat transfer to or from the surroundings will have a smaller effect on the \(ΔS_{surr}\). Conversely, if the Tsurr is low, the effect on ΔSsurr will be greater. (Atkins uses the analogy of the effect of a sneeze in a library compared to a crowded street; An American Chemistry General Chemistry text uses the analogy of giving $5 to a friend with $1000 compared to one with just $10.) Hence,
\begin{equation}
\Delta S_{\mathrm{surr}}=\frac{-\Delta \mathrm{H}_{\mathrm{sys}}}{\mathrm{T}}
\end{equation}
(Note: from a more rigorous thermodynamic approach, entropy can be determined from \begin{equation}
\mathrm{dS}=\frac{\mathrm{dq}_{\mathrm{rev}}}{\mathrm{T}}
\end{equation}
Once again,
\begin{equation}
\Delta S_{\text {tot }}=\Delta S_{\text {surr }}+\Delta S_{\text {sys }}
\end{equation}
\(ΔS_{tot}\) depends on both enthalpy changes in the system and entropy changes in the surroundings. Hence,
\begin{equation}
\Delta S_{\text {tot }}=\frac{-\Delta \mathrm{H}_{\mathrm{sys}}}{\mathrm{T}}+\Delta \mathrm{S}_{\mathrm{sys}}
\end{equation}
Multiplying both sides by \(-T\) gives
\begin{equation}
-\mathrm{T} \Delta \mathrm{S}_{\mathrm{tot}}=\Delta \mathrm{H}_{\mathrm{sys}}+\mathrm{T} \Delta \mathrm{S}_{\mathrm{sys}}
\end{equation}
The thermodynamic function Gibb's Free Energy, \(G\), can be defined as:
\begin{equation}
G=H-T S
\end{equation}
At constant \(T\) and \(P\),
\begin{equation}
\Delta G=\Delta H-T \Delta S
\end{equation}
Hence
\begin{equation}
\Delta G_{s y s}=\Delta H_{s y s}-T \Delta S_{s y s}=-T \Delta S_{t o t}
\end{equation}
Spontaneity is determined by \(ΔS_{tot}\) OR \(ΔG_{sys}\) since \(ΔS_{tot} = -ΔG_{sys}/T\). \(ΔG_{sys}\) is widely used in discussing spontaneity since it is a state function, depends only on the enthalpy and entropy changes in the system, and is negative (as is the potential energy change for a falling object) for all spontaneous processes.
The second law of thermodynamics can be succinctly stated: For any spontaneous process, the \(ΔS_{tot} > 0\). Unlike energy (from the First Law), entropy is not conserved.
Summary
(Summary written by Claude, Sonnet 4.6, Anthropic)
This chapter develops the thermodynamic framework needed to understand why biochemical reactions occur, to what extent they proceed, and how cells regulate them. Though the sheer number and complexity of cellular reactions may seem overwhelming, all are governed by the same physical principles that apply to any chemical system.
The extent of a reaction is described by its equilibrium constant, Keq. Reactions span a continuum from essentially irreversible (Keq >> 1, products strongly favored) to strongly favoring reactants (Keq << 1). The Keq reflects the intrinsic relative stability of reactants and products — a property independent of concentration — and is fixed for a given set of conditions. When a system at equilibrium is perturbed by changes in concentration, temperature, or removal of products, Le Châtelier's Principle predicts that the system will shift to counteract the perturbation. Cells exploit this principle constantly: by coupling reactions, removing products through further metabolism, or compartmentalizing pathways, they drive thermodynamically unfavorable conversions to completion.
The central thermodynamic quantity governing reaction spontaneity and direction is the Gibbs free energy change, ΔG, which integrates two distinct contributions: ΔG° (reflecting intrinsic molecular stability, equivalent to −RT ln Keq) and RT ln Q (reflecting the current concentration ratio of products to reactants). A reaction proceeds spontaneously in the direction that makes ΔG negative, continues until ΔG = 0 at equilibrium, and reverses if ΔG becomes positive. Because cellular reactions rarely occur at the biochemical standard state of 1 M, it is ΔG — not ΔG° — that determines reaction direction at any given moment. For biochemical systems, ΔG°' (defined at pH 7 rather than pH 0) is the appropriate standard-state reference.
The thermodynamic basis of spontaneity is fully captured in the Gibbs equation, ΔG = ΔH − TΔS. Enthalpy changes (ΔH) reflect the net difference in bond energies between reactants and products: exothermic reactions release heat (ΔH < 0) and are enthalpically favored, while endothermic reactions absorb heat (ΔH > 0) and are enthalpically disfavored. However, enthalpy alone does not determine spontaneity. Entropy (S), rigorously defined through Boltzmann's equation (S = k ln W) as a measure of the number of accessible microstates for energy and matter, provides the second essential contribution. Spontaneous processes tend to increase the dispersal of energy and matter — reflected in positive ΔS for processes such as dissolution, gas expansion, or the transition from ordered solids to disordered liquids and gases. The Second Law of Thermodynamics requires that the total entropy of the universe increases for any spontaneous process. Expressing this requirement in terms of system properties alone yields the Gibbs free energy function, making ΔG the practical and universally applied criterion for spontaneity in biochemical systems.