
52: DNA Restriction and Electrophoresis
- Page ID
- 3702
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Learning Objectives
- Learn how to cut DNA into fragments with enzymes.
- Load and separate DNA fragments by electrophoresis
- Analyze electrophoresis gels
DNA RESTRICTION ANALYSIS
In this experiment, DNA from the bacteriophage Lambda (48,502 base pairs in length) is cut with a variety of restriction enzymes and the resulting fragments are separated using gel electrophoresis. Three samples of Lambda (phage) DNA are incubated at 37º C, each with one of the 3 restriction endonuclease enzymes: Pst1, EcoRI, and HindIII. A fourth sample will be the negative control in that is will be incubated without any endonuclease. Each of the 3 enzymes recognizes a different sequence of bases on DNA called a pallindrome , and cuts within it at a specific site called a "restriction site."
 The DNA samples are then loaded into wells of an agarose gel and electrophoresed, along with loading dyes (see procedure below). An electrical field applied across the gel causes the DNA fragments in the samples to move from their origin (a sample well) through the gel matrix toward the positive electrode. Small DNA fragments migrate faster than larger ones, so restriction fragments of differing sizes separate into distinct bands during electrophoresis. The loading dyes are of 2 different sizes, corresponding to very small DNA fragments and very large DNA fragments. They can be seen as the electrophoresis progresses, and they form a 'bracket' in between which the DNA fragments are moving. Otherwise, one cannot tell how far the DNA fragments have moved through the agar. The characteristic number and pattern of bands produced by each restriction enzyme are made visible by staining with a compound that binds to the DNA molecule--- methylene blue.
The DNA samples are then loaded into wells of an agarose gel and electrophoresed, along with loading dyes (see procedure below). An electrical field applied across the gel causes the DNA fragments in the samples to move from their origin (a sample well) through the gel matrix toward the positive electrode. Small DNA fragments migrate faster than larger ones, so restriction fragments of differing sizes separate into distinct bands during electrophoresis. The loading dyes are of 2 different sizes, corresponding to very small DNA fragments and very large DNA fragments. They can be seen as the electrophoresis progresses, and they form a 'bracket' in between which the DNA fragments are moving. Otherwise, one cannot tell how far the DNA fragments have moved through the agar. The characteristic number and pattern of bands produced by each restriction enzyme are made visible by staining with a compound that binds to the DNA molecule--- methylene blue.
MATERIAL NEEDED
- restriction buffer
- loading dye
- ice
- 37º C water bath
- Lambda phage DNA
- TBE buffer mix
- restriction enzymes: Pst1, EcoR1, Hind111
- agarose to be poured (in 60º C water bath)
- reaction tubes
- 4 microtubes: clear, violet, green, orange
- staining tray
PROCEDURES
Use of the micropipetter
- Be sure that the amount set on the pipetter is correct.
- Place new tip firmly on micropipetter.
- Depress plunger to first stop, and hold this position. This step eliminates any air in tip.
- Dip tip into solution to be pipetted, and draw fluid into tip by releasing plunger. Always touch pipette tip to side of tube to dislodge any small amount stuck to tip. You now have a sample inside of your pipette tip.
- To expel sample, touch pipette tip to inside wall of tube into which you want to empty sample. This creates a capillary effect which will help draw fluid out of tip.
- Slowly depress plunger to first stop and then depress to second stop to blow out last bit of fluid in tip. DO NOT release plunger before removing tip from fluid in tube. Otherwise, it will suck fluid back into tip.
- When taking a sample, always check for air at the tip. If it is present, put the sample back and begin again.
Digest DNA with restriction endonucleases (keep all enzymes on ice)
- Label four 1.5ml tubes, in which you will perform restriction digestion: "P" for Pst1 enzyme, "E" for EcoRI enzyme, "H" for HindIII enzyme, and "L" for Lambda DNA uncut.
- Using table below, add reagents to each tube in this order: DNA, restriction buffer, water, and enzymes last (ask for them). Use a fresh pipette tip for each different reagent. The amounts are given in microliter (millionths of a liter).
- Pool and mix reagents by tapping the tube bottom on counter a couple of times (or use a microcentrifuge).
- Incubate all reaction tubes for 30 minutes at 37º C.
- The tubes will go into the freezer until the next period when the electrophoresis will be done.


Casting agarose gel
- Set gel-casting tray into the tray apparatus, screw tight, and insert well-forming comb into space marked with red line. There is a leveling bubble which can be used to level the tray (by turning knobs at bottom).
- Place tray FLAT where agarose can be poured and allowed to set UNDISTURBED.
- Carefully pour 40 ml of agarose solution (liquified in 60º C water bath) into casting tray. Use a toothpick to move any bubbles to edges (this must be done BEFORE gel hardens).
- Gel will solidify within 20 minute. Do NOT move tray while agarose is solidifying.
- Gently remove comb, pulling it straight up and taking care not to rip wells.
- When solidified, remove the gel tray from the gel-casting tray and place on platform of electrophoresis box, so that comb is at negative (BLACK) cathode end. The - charged DNA fragments will migrate towards the + anode end.
- Fill box with TBE buffer, to level that just covers entire surface of gel by about 2 mm.
- Make certain that sample wells left by comb are completely submerged by buffer.
- The gel is now ready to load with DNA.
Loading gel with DNA
- Remove the 4 tubes from the fridge and pulse spin the tubes in a centrifuge or tap the tubes firmly down on the table top so that all contents go down to the bottom of the tube.
- Add 2 µl loading dye to each reaction tube and tap contents of tube on table top.
- Use pipette to load contents of each reaction tube into a separate well in gel (total of 4 wells). Set your micropipetter on 12 µl (that should be the total contents in the tube).
- You will need to remember the order of your tubes since there is NO way that the gel can be marked. Use a clean pipette tip for each different tube.
- Steady pipette over well using 2 hands.
- Expel any air first from pipette tip.
- Dip pipette tip through buffer, positioned over the well, and slowly expel the mixture (do not punch thru bottom of gel).
- The loading dye contains sucrose which is heavier than the DNA. It weighs the mixture down so it will sink into the bottom of the well.
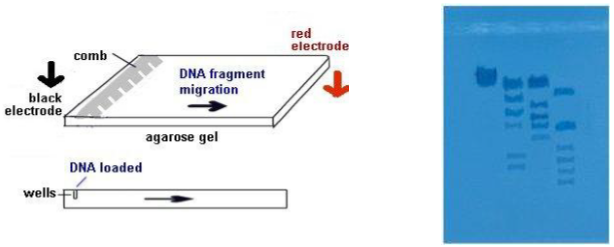
Electrophoresis
- The electrophoresis chamber top is placed on the chamber, the electrodes connected to power supply--anode to anode (red-red) and cathode to cathode (black-black).
- Power supply is turned on and voltage set---120 V. The higher the voltage, the faster the electrophoresis time. In a few minutes, you should begin to notice the loading dye moving through the gel toward the + pole (anode). We will let it run for about 1 hour.
- The loading dye will resolve into 2 bands of color. The faster-moving, purplish band is the dye bromophenol blue, the slower-moving, aqua band is xylene cyanol . Bromophenol blue migrates through the gel at the same rate as a DNA fragment approximately 300 base pairs long. Xylene cyanol migrates at the a rate equivalent to approximately 2,000 base pairs long.
- After 1 hour, the bromophenol band should be nearing the end of the gel. Turn off the power supply, disconnect the leads, and remove the top of chamber.
- Carefully remove the casting tray and keep it horizontal until you are ready to put into the plastic container. Slide gel easily into staining tray labeled with your group name.
- Add enough methylene blue DNA stain to cover the gel and place cover on it. They will sit in the dye overnight.
- The gels will be washed a couple of times in distilled water (standing for 10-20 min), and will refrigerate until next lab period when we will look at them.
- The gels can then be placed on gel support film, which binds the gel and dehydrates it, if your instructor so chooses.
INTERPRETATION
 Examine your stained gel on a light box, overhead projector, or a UV box. Which restriction enzyme produced the most restriction sites on the lambda DNA?
Examine your stained gel on a light box, overhead projector, or a UV box. Which restriction enzyme produced the most restriction sites on the lambda DNA?
The HindIII digest of lambda DNA yields at least 6 fragments suitable for use as molecular weight standards for gel electrophoresis. This is called a ladder. After the sample is ran, the unknown fragments can be compared with the ladder fragments to determine the approximate size of the unknown DNA bands by how they match up to the known bands of the ladder.

QUESTIONS
- What is the source of the DNA being used in this exercise?
- The speed of the DNA fragment migration correlates directly with the _____ of the fragment.
- Why use 3 different restriction enzymes to cut DNA?
- How can you account for differences in band separation and intensity between your gel and the ideal gel?
- Small restriction fragments of nearly the same length will appear as a single band on this gel, even though it may be run to the very end. Why?
Contributors and Attributions
-
Jackie Reynolds, Professor of Biology (Richland College)