
11: Bacterial Numbers
- Page ID
- 3485
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Learning Objectives
- Correlate absorbance value for a bacterial suspension with an accurate bacterial count.
- Become proficient at dilutions.
- Become proficient at performing a standard plate count and determining bacterial counts in a sample.
Many studies require the quantitative determination of bacterial populations. The two most widely used methods for determining bacterial numbers are the standard, or viable, plate count method and spectrophotometric (turbidimetric) analysis. Although the two methods are somewhat similar in the results they yield, there are distinct differences. For example, the standard plate count method is an indirect measurement of cell density and reveals information related only to live bacteria. The spectrophotometric analysis is based on turbidity and indirectly measures all bacteria (cell biomass), dead and alive.
The standard plate count method consists of diluting a sample with sterile saline or phosphate buffer diluent until the bacteria are dilute enough to count accurately. That is, the final plates in the series should have between 30 and 300 colonies. Fewer than 30 colonies are not acceptable for statistical reasons (too few may not be representative of the sample), and more than 300 colonies on a plate are likely to produce colonies too close to each other to be distinguished as distinct colony-forming units (CFUs). The assumption is that each viable bacterial cell is separate from all others and will develop into a single discrete colony (CFU). Thus, the number of colonies should give the number of bacteria that can grow under the incubation conditions employed. A wide series of dilutions (e.g., 10-4 to 10-10) is normally plated because the exact number of bacteria is usually unknown. Greater accuracy is achieved by plating duplicates or triplicates of each dilution, although we will not be doing that in this exercise.
Increased turbidity in a culture is another index of bacterial growth and cell numbers (biomass). By using a spectrophotometer, the amount of transmitted light decreases as the cell population increases. The transmitted light is converted to electrical energy, and this is indicated on a galvanometer. The reading, called absorbance or optical density, indirectly reflects the number of bacteria. This method is faster than the standard plate count but is limited because sensitivity is restricted to bacterial suspensions of 107cells or greater. The procedure for the spectrophotometer use is at the end of this exercise.
Why Is E. coli used in this exercise? When working with large numbers and a short time frame, one of the most reliable microorganisms is one that has been used in previous experiments, namely, Escherichia coli. E. coli has a generation time at 37ºC of 20 minutes. Thus, it reproduces very rapidly and is easy to quantify (i.e., the number (biomass) of viable E. coli cells in a bacterial culture can be easily determined by spectrophotometry).
MATERIALS NEEDED: per table (exercise performed by table)
- 24-hour 10ml nutrient broth culture of Escherichia coli
- 4 sterile 99ml saline bottles
- 1ml and 5ml pipets with pi-pumps (green for 5ml, blue for 1ml)
- 6 petri plates
- 6 agar pour tubes of nutrient agar (plate count agar)
- 48ºC to 50ºC water bath
- 6 micro-cuvettes and rack
- 1 SpectroVis micro-cuvette holder
- computer
- 4 tubes of 5ml nutrient broths
THE PROCEDURES
STANDARD PLATE COUNT
Note
BE SURE TO SAVE YOUR ORIGINAL TUBE OF E.COLI FOR THE NEXT SECTION!
Your agar deeps should already be liquefied and sitting in a water bath. If not, you will have to boil them. When the deeps are liquefied, they can be placed into the water bath to cool so that the pours can be made without killing the bacteria. Coolingtakes about 20 minutes.
- Label the bottom of six petri plates 1-6. Label four tubes of saline 10-2, 10-4, 10-6, and 10-8.
- Using aseptic technique, the initial dilution is made by transferring 1 ml of E. coli sample to a 99ml sterile saline blank (figure below. This is a 1/100 or 10-2 dilution.
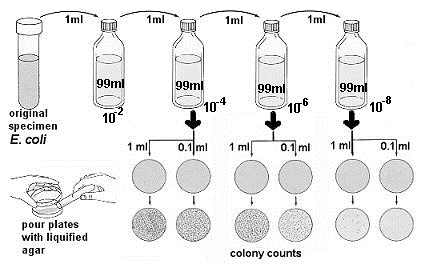 Immediately after the 10-2 dilution has been shaken, uncap it and aseptically transfer 1 ml to a second 99 ml saline blank. Since this is a 10-2 dilution, this second blank represents a 10-4 dilution of the original sample.
Immediately after the 10-2 dilution has been shaken, uncap it and aseptically transfer 1 ml to a second 99 ml saline blank. Since this is a 10-2 dilution, this second blank represents a 10-4 dilution of the original sample.- Shake the 10-4 dilution vigorously and transfer 1 ml to the third 99 ml blank. This third dilution represents a 10-6 dilution of the original sample. Repeat the process once more to produce a 10-8 dilution.
- Shake the 10-4 dilution again and aseptically transfer 1.0 ml to one petri plate and 0.1 ml to another petri plate. Do the same for the 10-6 and the 10-8 dilutions.
- Remove one agar pour tube from the 48º to 50ºC water bath. Carefully remove the cover from the 10-4 petri plate and aseptically pour the agar into it. The agar and sample are immediately mixed gently moving the plate in a figure-eight motion or a circular motion while it rests on the tabletop. Repeat this process for the remaining five plates.
- After the pour plates have cooled and the agar has hardened, they are inverted and incubated at 25ºC for 48 hours or 37ºC for 24 hours.
- At the end of the incubation period, select all of the petri plates containing between 30 and 300 colonies. Plates with more than 300 colonies cannot be counted and are designated too many to count (TMTC). Plates with fewer than 30 colonies are designated too few to count (TFTC). Count the colonies on each plate. A Quebec colony counter should be used.
- Calculate the number of bacteria (CFU) per milliliter or gram of sample by dividing the number of colonies by the dilution factor multiplied by the amount of specimen added to liquefied agar.
number of colonies (CFUs) = # of bacteria/ ml
dilution X amount plated
- Record your results.
TURBIDIMETRY DETERMINATION OF BACTERIAL NUMBERS
Note
This section does not have to be done aseptically!
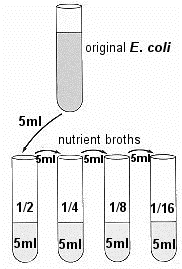 Place the ORIGINAL tube of E. coli and four tubes of the sterile NB in a test-tube rack. Each tube of NB contains 5 ml of sterile broth. Use four of these tubes (tubes 2 to 5) of broth to make four serial dilutions of the culture.
Place the ORIGINAL tube of E. coli and four tubes of the sterile NB in a test-tube rack. Each tube of NB contains 5 ml of sterile broth. Use four of these tubes (tubes 2 to 5) of broth to make four serial dilutions of the culture. - Transfer 5ml of E. coli to the first tube of NB, thoroughly mixing the tube afterwards. Transfer 5ml from that tube to the next tube, and so on until the last of the 4 tubes has 5ml added to it. These tubes will be 1/2, 1/4, 1/8, and 1/16 dilutions.
- The directions for spectrophotometer use are BELOW.
- Record your values, along with the dilutions that they came from. Using the plate count data, calculate the colony-forming units per milliliter for each dilution.
DATA COLLECTION
| dilutions | absorbance (X) | # of bacteria (Y) |
| original E. coli | ||
| 1/2 | ||
| 1/4 | ||
| 1/8 | ||
| 1/16 |
- Fill in your absorbance values for the 5 tubes read in the spectrophotometer.
- Calculate the number of bacteria in the original tube of E. coli, and place that value in the top right cell of the table. This is done AFTER THE PLATES HAVE INCUBATED.
- Calculate the approximate numbers of bacteria in the 1/2, 1/4, 1/8, and 1/16 by halving the number in the cell above.
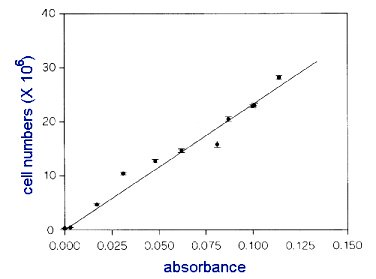
- Plot these 5 coordinates on a graph, using EXCEL software (it is available in the computer labs). The DIRECTIONS on howto use the software is at end of exercise.
- Here is an example of a graph.
Note
YOU MUST HAVE EQUAL INTERVALS ALONG BOTH X AXIS AND Y AXIS.
QUESTIONS
- DATA COLLECTION:
| dilutions | absorbance (X) | # of bacteria (Y) |
| original E. coli | ||
| 1/2 | ||
| 1/4 | ||
| 1/8 | ||
| 1/16 |
- Why do a standard plate count when running turbidity values the first time?
- If you have a graph for E. coli, can it also be used for another bacterium like Staph?
- How is transmission different from absorbance?
- Give the formula for determining bacterial counts.
- Give the bacterial count per milliliter of E. coli suspension in the original culture tube.
Using the Vernier SprectroVIS Spectrophotometer to measure absorbances as a function of concentration
Note
UNPLUG AND RE-PLUG THE SPECTROVIS USB INTO THE COMPUTER BEFORE BEGINNING. IT GIVES A BETTER READING.
I. Calibration
 There is no on/off switch for the spectrophotometer; just make sure that the USB cable is connected to the computer.
There is no on/off switch for the spectrophotometer; just make sure that the USB cable is connected to the computer.- The SpectroVisis plugged into the computer BEFORE opening the software.
- Double click on the Logger Pro icon on the computer desktop. When you open the Logger Pro 3.8.2, the light will be turned on.
- Fill a cuvette (about ¾ full) with the blank solution (tryptic soy broth for this experiment).
- Insert the cuvette in the sample chamber, making sure that the clear side of the cuvette is in the path of the light.
- On the top menu, click on Experiment. Scroll down to Calibrate and select Spectrometer 1.
- Wait 60 seconds for the lamp to warm up.
- Click on Finish Calibration and then OK. The spectrophotometer is now calibrated.
II. Setting the machine for data capture –BACTERIAL NUMBERS experiment
- On the top bar, find the Configure Spectrophotometer icon.
- Select Absorbance vs. Concentration as the Collection Mode.
- Below the concentration menu, select Individual Wavelengths.
- Under the column showing a list of wavelengths, click on Clear Selections and the select the wavelength closest to 600nm as the desired wavelength.
- A graph will appear with concentration on the X-axis and absorbance at 600 on the Y-axis.
- REMOVE the blank cuvette.
III. Reading absorbance of the BACTERIAL DILUTION SAMPLES
- With the cuvette containing the original E.coli solution (1st dilution) in the chamber, click on Collect on the top menu.
- Wait until a red dot appears on the screen and click on Keep on the top bar.
- A menu appears, asking for the concentration of the sample. Use 1 for this dilution.
- Click on Keep with the 1/2 dilution of bacteria, enter .5 for the concentration.
- Click on Keep with the 1/4 dilution of bacteria, entering .25 for the concentration.
- Click on Keep with the 1/8 dilution of bacteria, entering .125 for the concentration.
- Click on Keep with the 1/16 dilution of bacteria, entering .0625 for the concentration.
- Click Stop on the top bar menu.
Plotting Data Using Microsoft Excel
Plotting Best-Fit Lines of Absorbance vs. Numbers data
- Open the Microsoft Excel program. You will see a grid. Each box in the grid is called a cell. Each cell has an “address” made up of a letter indicating the column the cell is in and a number indicating the row the cell is in, e.g. the upper left cell is A1.
- Double click on cell A1 to start entering information. Using the diagram on this next page as a reference, enter the data of bacterial numbers and absorbance readings of your table.
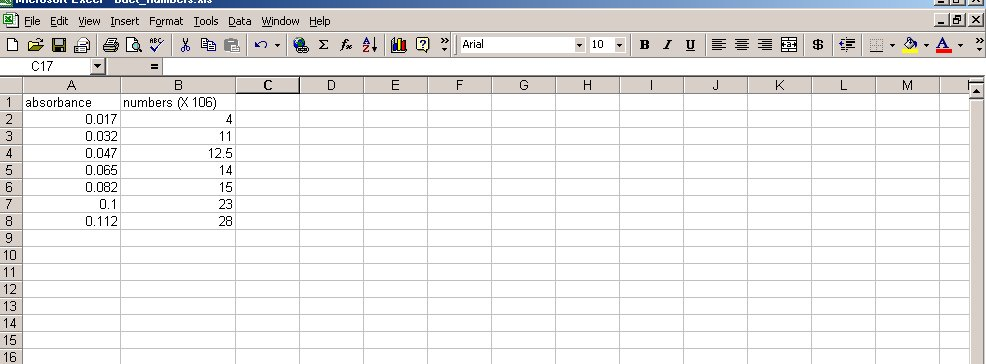
- Click on cell A2 with the left mouse button (LMB) and drag the mouse to highlight the area containing all the data points (go down to cell B8). The screen should look like this:
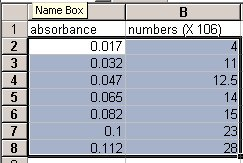
MICROSOFT OFFICE 2007
Note
DIRECTIONS DIFFER, DEPENDING ON WHETHER YOU HAVE OFFICE 2003 OR 2007
- On the top bar, click on Insert and select Scatter (in Chart Area). Select first choice, Scatter with only markers (unconnected dots).
- Click on the graph. Look for “Chart Tools” on the top bar. Pick Layout. Click on Chart Title, select “Above Chart.” Type in the title of the graph.
- In Layout………Click on Axis Titles (on the top bar). Select Primary Horizontal. Select Title Below Axis. Type in the title of the axis. e.g. Absorbance
- Click on Axis Titles again. Select Primary Vertical. Select Rotated Title. Type in the title of the axis. e.g. Bacterial Numbers. Do NOT FORGET to include X 10 (whatever exponent you are using), e.g. 106. To make a superscript exponent for a number,type 106, and select the 6. Click on FONT, choose superscript and press OK.
- Right click on a data point. Click on “Add Trendline”. On the next screen,be sure that LINEAR is checked and then click on Display Equation.
- Click on Gridlines. Make sure you are showing horizontal and vertical lines. Click on Primary Horizontal and choose Major and Minor gridlines. Click on Primary Vertical and choose Major and Minor gridlines.
- Delete each “Linear Series” from the legend box by clicking on each and on delete. Make sure that there are “dots” ONLY around “Linear Series” box and not around the Legend Box, otherwise you will delete the whole legend box.
- You might want to select graph by clicking on it, to move it around the page. To print the graph FULL-SIZE on page, click to select graph and click on print. If you click on Print without doing the above, you will get the X and Y value table along with the graph, but your graph will be tiny (below).
- Before proceeding any further, you want to be sure that all your work is saved.
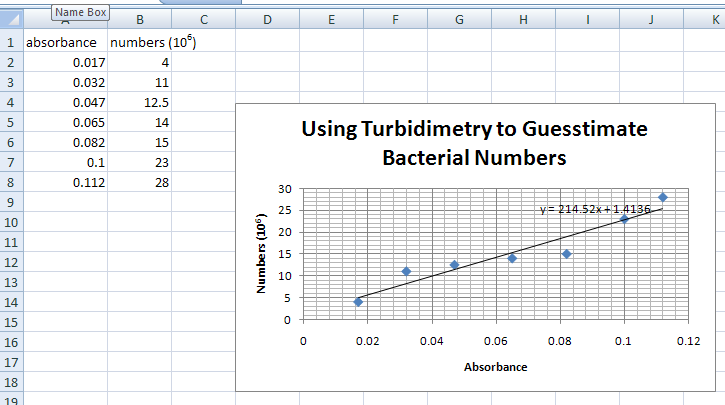
THE GRAPH SHOULD LOOK LIKE THE PICTURE ABOVE, BUT WITH A LINE AND THE SLOPE FORMULA.
MICROSOFT OFFICE 2003
- Once the data is highlighted, look on the upper toolbar and click on “Insert”, then on “Chart”. A box with graph options will appear. Select “XY(Scatter)” and click on “Next”. (Leave the chart sub-type alone: the top chart should be highlighted.)
Your data will appear already plotted on an x-y axis system. Now you will learn how to set-up the axes, write the title of the graph and write the legend. You will also command the computer to draw best-fit lines and calculate the slope of each plot. You can also choose to make some cosmetic touches (changing the size of fonts, colors of the plot etc.…) on your graph before printing.
- Find “Series” towards the top of the graph and click on it. “Series1” should appear highlighted. To the right of this there is an empty space for you to write the NAME of this plot: click on this empty box and type “Absorbance vs. Bacterial Numbers”.
- Click on “Next”. Another box will appear with options to modify titles on the graph.
a. Make sure that in the window “Chart Title” your graph is titled (e.g. Using Turbidemetry to Guesstimate Bacterial Numbers”).
b. Click on space below Value(X)Axis and type: Absorbance
c. Click on space below Value(Y)Axis and type: Bacterial Numbers (X 106)
d. Click on “Axes” (upper left of graph) and make sure that Value(X) Axis and Value(Y) Axis have a √ .
e. Click on “Gridlines” and add lines. Click on “Major Gridlines” on the x-axis and y-axis.)
f. Click on “Legend” and choose the location of the legend (I chose bottom).
- Click on “Next” click on the button titled Place chart as object in sheet 1 (NOT as new sheet). Click on “Finish”.
Your graph will appear on the screen as shown below. You can alter its size by clicking the mouse button on the black squares around the graph and dragging the mouse with the mouse button pressed down.
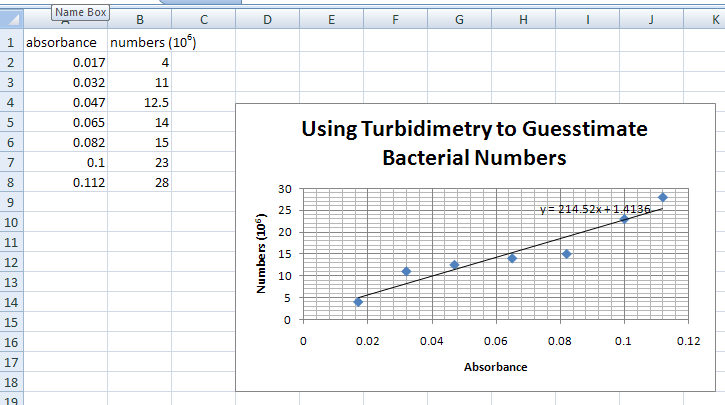
- Now you are going to add the line that goes through the data points (trendline). Click on the graph to make sure that the black squares around the graph are there.
Move the pointer to any of the data points of a particular series (any Δ, for example). As you do this, information about this data point will appear on the screen. Press the RIGHT mouse button. A box with choices appears. Select and click on “Add Trendline”. Another box will appear. Select Linear (should already be highlighted). Before clicking OK, go to “Options” on the top of the box. Click on “Display equation on chart”. Click on “OK”. The graph will show a best-fit line. On the graph you should see an equation which gives you the value of the slope for the line).
THE GRAPH SHOULD LOOK LIKE THE PICTURE ABOVE, BUT WITH A LINE AND THE SLOPE FORMULA.
- Cosmetic changes for graphs. You can change font size, color, etc. if you like. However, I will not get into those directions here.
- Go to the top toolbar. Click on “File”, “Print Preview” and check if the graph is ready to print. If it is ready, click on “Print” and “OK”.
- Before proceeding any further, you want to be sure that all your work is saved.
Contributors and Attributions
Jackie Reynolds, Professor of Biology (Richland College)