
2: SPOC II- Genomes
- Page ID
- 163000

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Distinguish between genes, chromosomes, and genomes.
- Describe the range in the size of genomes and numbers of genes between species.
- Distinguish between SNPs and STRs as variations in DNA sequence.
- List applications of the field of genomics.
What is a genome?
The complete set of an organism's genetic information is the genome.
Variation exists when sequences of bases differ between chromosomes. Some examples of types of variation include:
- differences between the base sequence of homologous chromosomes in an individual
- differences between the base sequence of chromosomes in two individuals
- differences in base sequences between two different species
- differences in numbers of chromosomes between species
- differences in arrangement of similar sequences between species (synteny)
- differences in base sequence individuals with and without a disease or trait
....and more
The study of sequences at the whole genome level is known as genomics.
Why study genomes?
Understanding genomes has a variety of applications including:
- comparing species to understand gene function and evolution
- understanding health differences between individuals
- performing research into gene function using model organisms
- identifying individuals or species
Genome sequencing projects
The first organismal genome to be sequenced was the prokaryote H. influenzae in 1995 (Fleischmann et al, 1995).
Determining the sequence of the entire human genome took more than two decades from its initial official start in 1990 to the draft sequence published in 2001 and the refinement of that draft sequence over the next several years (review key dates in the timeline here: https://www.genome.gov/about-nhgri/Brief-History-Timeline#one). Sequences produced by genome projects are referred to as reference genomes.
The results of human genome sequencing efforts were published at the same time in Nature (results of the International Human Genome Sequencing Consortium) and Science (by Craig Venter and colleagues with Celera Genomics) in February 2001. These two groups had taken somewhat different approaches to sequencing efforts.
However, there were still some "gaps" in the sequence. For example, regions that are highly repetitive or similar to other chromosomes are difficult to sequence precisely. The next-generation sequencing techniques described previously have facilitated obtaining complete chromosome sequences for some of these missing regions. Recently, the complete sequence of an X chromosome from "telomere-to-telomere" was published (Miga et al., 2020), followed by a complete assembly of chromosome 8 (Logsdon et al., 2021).
Exercise \(\PageIndex{1}\)
Whose genome was the first sequenced?
For the Celera project, refer to the Sources of DNA and Sequencing Methods section of this paper https://science.sciencemag.org/content/291/5507/1304.long.
For the International consortium project, the sample collection is reported in this paper https://www.nature.com/articles/35057062.
- Answer
-
Celera enrolled 21 donors initially, but only 5 of these were used for the complete sequencing. What factors determined which samples were selected?
For the International consortium, volunteer donors were solicited from areas near research labs. After sample collection and processing all identifying information was removed. Many more samples were collected than were actually used. So truly, we don't know whose genome is included!
How big are genomes? And does it matter?
Genomes vary in size and complexity. Bacterial genomes are the smallest in numbers of base pairs, followed by fungi and algae. Beyond these organisms, genome size does not necessarily increase with complexity. Plants have some of the largest genomes (often because they have more than two sets of chromosomes). However, regardless of the total genome size, most organisms does not have much more than 25,000 genes, so total size does not necessarily increase gene number.

Figure \(\PageIndex{1}\): Genome sizes (in base pairs) vary by organism. (CC:BY-SA Abizar via Wikipedia Commons)
Remember that much genome research is funded by grants from governments and is made publicly accessible. Some databases that maintain genome information are:
- National Center for Biotechnology Information (NCBI) Genome Data Viewer https://www.ncbi.nlm.nih.gov/gdv/
- European Molecular Biology Laboratory's European Bioinformatics Institute Ensembl https://www.ensembl.org/index.html
- University of Californai Santa Cruz Genomics Institute https://genome.ucsc.edu/
Variation: SNPs and STRs
Genome sequencing projects reveal the reference genome that is largely shared between species or individuals, but they also reveal variation. Although other types of variation exist, two major classes include single nucleotide polymorphisms (SNPs) and short tandem repeats (STRs). SNPs are positions along chromosomes that can vary in the nucleotide at that position. STRs are repeats of a few nucleotides that can vary in the number of repeats.
The Human Genome Viewer
The Human Genome Project included much more than just the sequence of one human's genome. Since the project started, data has been added from a huge number of humans and many annotations have been added. There is so much data available that specialized viewers have been developed to allow people to explore these massive datasets. One of these viewers, The Genome Data Viewer from the National Institutes of Health, is free to all users.
The remaining Graded Problems in this section will show you how to use the Genome Data Viewer (GDV). We will also use this software in class.
You can access the Genome Data Viewer from this link.
The video below may help with the steps below:
1) Click on the link above to load the GDV. You will see something like this:

2) You type the name (or other relevant information) into the Search Bar and click the arrow (or press the Return key). For this problem, enter "HBA1" and press return. You should see something like this (if you don't see the same list in the "Genes" tab, check to be sure you entered "HBA1" (no quotes) into the search bar). If you hover the mouse over the HBA1 entry, a window will pop up as shown below:

The pop-up contains a lot of useful information. In this case, it tells us that the gene is ("Description") the hemoglobin subunit alpha 1 - one of the two components of the protein hemoglobin that carries oxygen in the blood.
For our purposes, it is also useful to know which chromosome carries this gene. This is in the "Location" entry. The chromosome number or name is preceeded by "Chr" which is short for "Chromosome".
3) Click on the "HBA1" entry at the top of the list in the Genes tab. You should see something like this. Be sure that the "Gene" tag in the top blue part (arrow) says "HBA1"; if it doesn't, try clicking on the HBA1 entry again in the list in the Genes tab.

Next, look at the panel to the right. It contains a wealth of data about this gene.
The most important part of this is the top green line (in the figure above, it is labeled "NM_000558.5").
- The black or white arrows in the gene show the direction of transcription (left to right or right to left)
- The thick parts are exons, the thin parts are introns.
- The darker parts of the exons indicate the coding region (the part that encodes the amino acids in the protein).
If you hover the mouse over the top green line, you should see something like this:

There are several important parts to this:
- Span on ... - this gives the size of the gene in nucleotides (nt). In this case, it is 843 nucleotides long.
- Protein length - gives the size of the protein in amino acids (aa).
- Exon - says that there are 3 exons.
4) To see more detail about the gene and to scroll along the chromosome, you use these controls:

- the arrows scroll left and right
- the "-" in the magnifying glass zooms out
- the "+" in the other magnifying glass zooms in
The next step is to zoom in on the gene and explore some particular mutations.
Click on the "+" in the controls until you see this in the "Cited Variations" part of the window - it is right below the last of the green lines.

Each of the red squares corresponds to a different mutation in the gene.
The red square aligns vertically with its location in the gene.
If you hover the mouse over a red square, you'll see something like this (the details will differ depending on which one you choose):
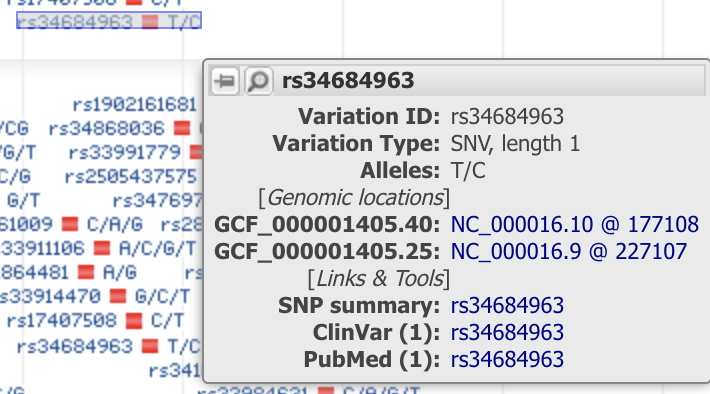
There are three important parts to this panel:
- Variation Type - in this case, SNV (single nucleotide variation) of length 1. This means "this variation changes only one base in the DNA"
- Alleles - this gives the specific change in the format "(original base)/(mutant form or forms)"
- PubMed - this is a link to any scientific publications about this variant (we'll use this in class).
References
Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496-512. doi:10.1126/science.7542800
International Human Genome Sequencing Consortium., Whitehead Institute for Biomedical Research, Center for Genome Research:., Lander, E. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). https://doi.org/10.1038/35057062
Logsdon, G.A., Vollger, M.R., Hsieh, P. et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021). https://doi.org/10.1038/s41586-021-03420-7
Miga, K.H., Koren, S., Rhie, A. et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). https://doi.org/10.1038/s41586-020-2547-7
Milo, Ron and Philips, Rob. Cell Biology by the Numbers. Garland Science, 2015.